Ofloxacine
- Product NameOfloxacine
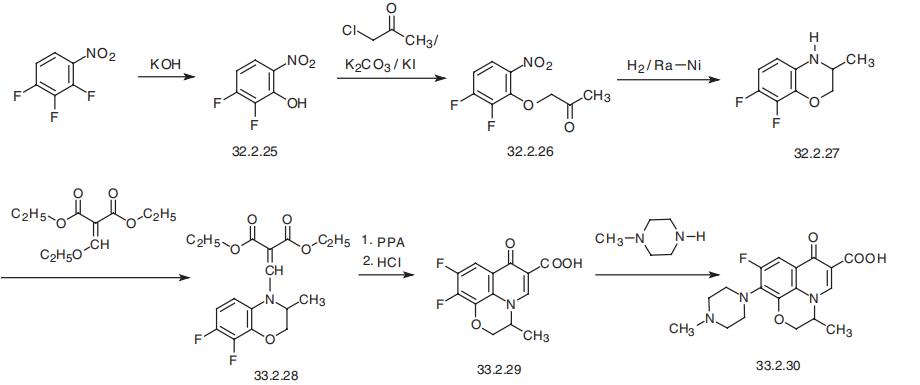
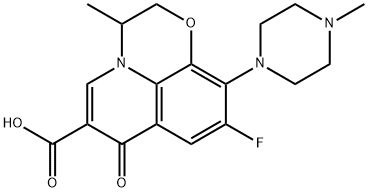
- CAS82419-36-1
- MFC18H20FN3O4
- MW361.37
- EINECS680-263-1
- MOL File82419-36-1.mol
Chemical Properties
| Melting point | 270-2750C |
| Boiling point | 571.5±50.0 °C(Predicted) |
| Density | 1.2688 (estimate) |
| storage temp. | Keep in dark place,Sealed in dry,Store in freezer, under -20°C |
| solubility | 1 M NaOH: soluble50mg/mL |
| pka | 5.19±0.40(Predicted) |
| form | Solid |
| color | Colorless needles from ethanol |
| biological source | synthetic |
| Water Solubility | Soluble in acetic acid or water. Slightly soluble in methanol |
| λmax | 326nm(H2O)(lit.) |
| Merck | 14,6771 |
| BCS Class | 1 |
| CAS DataBase Reference | 82419-36-1(CAS DataBase Reference) |
| EPA Substance Registry System | Ofloxacin (82419-36-1) |
Safety Information
| Hazard Codes | Xn,Xi |
| Risk Statements | 22-42/43-68-36/37/38 |
| Safety Statements | 26-36/37/39-24/25-22-37/39-60 |
| WGK Germany | 3 |
| RTECS | UU8815550 |
| HS Code | 29349990 |
| Hazardous Substances Data | 82419-36-1(Hazardous Substances Data) |
| Toxicity | LD50 in male, female mice, male, female rats (mg/kg): 5450, 5290, 3590, 3750 orally; 208, 233, 273, 276 i.v.; >10000, >10000, 7070, 9000 s.c. (Ohno) |