Propiconazole: Uses, Pharmacology and Preparation
General Description
Meloxicam is a nonsteroidal anti-inflammatory drug (NSAID) used to relieve various types of pain, including pain caused by musculoskeletal conditions, osteoarthritis, and rheumatoid arthritis. With a longer half-life than most other NSAIDS, it is a favorable option for those who require once-daily dosing. Meloxicam is available in oral, transdermal, and intravenous formulations. It is a preferential COX-2 inhibitor, purportedly reducing the risk of adverse gastrointestinal tract effects, however, this is a topic of controversy. Meloxicam is indicated for the symptomatic treatment of arthritis and osteoarthritis. In addition, it is indicated for the pauciarticular and polyarticular course of Juvenile Rheumatoid Arthritis (JRA) in patients aged 2 years old or above. Off-label uses include the treatment of dental or post-surgical pain. In addition to the above, meloxicam has also been studied in the treatment of neuropathic pain. Meloxicam was launched in 1996 by Boehringer Ingelheim for the treatment of osteoarthritis and rheumatoid arthritis. The active ingredient of the drug product is polymorph Form I. Further known crystalline forms are Form II,7 Form III,7 Form IV (zwitterionic form) and Form V.1-5

Figure 1. Properties of Meloxicam oral tablet
Mechanism of action
Meloxicam inhibits prostaglandin synthetase (cylooxygenase 1 and 2) enzymes leading to a decreased synthesis of prostaglandins, which normally mediate painful inflammatory symptoms. As prostaglandins sensitize neuronal pain receptors, inhibition of their synthesis leads to analgesic and inflammatory effects. Meloxicam preferentially inhibits COX-2, but also exerts some activity against COX-1, causing gastrointestinal irritation. Meloxicam concentrations in synovial fluid range from 40% to 50% of those in plasma. The free fraction in synovial fluid is 2.5 times higher than in plasma, due to the lower albumin content in synovial fluid as compared to plasma. The significance of this penetration is unknown, but it may account for the fact that it performs exceptionally well in treatment of arthritis in animal models.6
Adverse effects
Meloxicam use can result in gastrointestinal, toxicity and bleeding, headaches, rash, and very dark or black stool (a sign of intestinal bleeding). It has fewer gastrointestinal side effects than diclofenac, piroxicam, naproxen, and perhaps all other NSAIDs which are not COX-2 selective.
Cardiovascular
Like other NSAIDs, its use is associated with an increased risk of cardiovascular events such as heart attack and stroke. Although meloxicam inhibits formation of thromboxane A, it does not appear to do so at levels that would interfere with platelet function. A pooled analysis of randomized, controlled studies of meloxicam therapy of up to 60 days duration found that meloxicam was associated with a statistically significantly lower number of thromboembolic complications than the NSAID diclofenac (0.2% versus 0.8% respectively) but a similar incidence of thromboembolic events to naproxen and piroxicam.7
Gastrointestinal
NSAIDs cause and increase the risk of serious gastrointestinal adverse events including bleeding, ulceration, and perforation of the stomach or intestines, which can be fatal. Elderly patients are at greater risk for serious gastrointestinal events.8
Pharmacodynamics
Meloxicam is an anti-inflammatory, analgesic analgesic with antipyretic effects in fever. Prostaglandins are substances that contribute to inflammation. This drug also exerts preferential actions against COX-2, which may reduce the possible gastrointestinal effects of this drug. In humans, meloxicam has demonstrated the ability to decrease erythrocyte sedimentation rate(ESR) in patients with rheumatoid arthritis, and to decrease ESR, C-reactive protein (CRP), as well as aquaporin-1 expression. As with other NSAIDS, prolonged use of meloxicum can result in renal or cardiovascular impairment or thrombotic cardiovascular events.
As meloxicam preferentially inhibits COX-2, it is thought to cause less gastrointestinal irritation compared to other NSAIDS. Despite this, it still carries a risk of gastric inflammation, bleeding and ulceration. In one study, patients on meloxicam suffered from gastrointestinal symptoms at a rate of 13% compared to 19% of those on diclofenac. GI events were found to be less severe in the meloxicam-treated patients.1-3
Pharmacokinetics
Absorption
The absolute bioavailability oral capsules after a dose was 89% in one pharmacokinetic study. Cmax was reached 5–6 hours after administration of a single dose given after the first meal of the day. The Cmax doubled when the drug was administered in the fasting state. Despite this, meloxicam can be taken without regard to food, unlike many other NSAIDS. Meloxicam formulated for instillation with bupivacaine produced varied systemic measures following a single dose of varying strength. In patients undergoing bunionectomy, 1.8 mg of meloxicam produced a Cmax of 26 ± 14 ng/mL, a median Tmaxof 18 h, and an AUC∞ of 2079 ± 1631 ng*h/mL. For a 9 mg dose used in herniorrhaphy, the corresponding values were 225 ± 96 ng/mL, 54 h, and the AUC∞ was not reported. Lastly, a 12 mg dose used in total knee arthroplasty produced values of 275 ± 134 ng/mL, 36 h, and 25,673 ± 17,666 ng*h/mL.9
Volume of distribution
The volume of distribution of meloxicam is 10-15L. Because of its high binding to albumin, it is likely to be distributed in highly perfused tissues, such as the liver and kidney. Meloxicam concentrations in synovial fluid, measured after an oral dose, is estimated at 40% to 50% of the concentrations measured in the plasma.10
Metabolism
Meloxicam is almost completely metabolized. CYP2C9 is the main enzyme responsible for the metabolism of meloxicam with minor contributions from CYP3A4. Meloxicam has 4 major metabolites with no activity determined. About 60% of the ingested dose is metabolized to 5'-carboxy meloxicam from hepatic cytochrome enzyme oxidation of an intermediate metabolite, 5'-hydroxymethylmeloxicam. Two other metabolites are likely produced via peroxidation.11
Excretion
Meloxicam is mainly eliminated through metabolism. Its metabolites are cleared through renal and fecal elimination. Less than <0.25% of a dose is eliminated in the urine as unchanged drug. About 1.6% of the parent drug is excreted in the feces.1
Half-life
The half-life of meloxicam is approximately 20 hours, which is considerably longer than most other NSAIDS. It can therefore be dosed without the need for slow-release formulations. Meloxicam applied together with bupivacaine for postsurgical analgesia had a median half-life of 33-42 hours, depending on dose and application site.9
Clearance
After an oral dose, the total clearance of meloxicam is 0.42–0.48 L/h. The FDA label indicates a plasma clearance from 7 to 9 mL/min. No dose changes are required in mild to moderate renal or hepatic impairment. The use of meloxicam in patients with severe renal or hepatic impairment has not been studied. FDA prescribing information advises against it.12
Toxicity
The oral LD50 in rats is 98 mg/kg. Signs and symptoms of overdose with meloxicam may include shallow breathing, seizure, decreased urine output, gastrointestinal irritation, nausea, vomiting, gastrointestinal bleeding, and black tarry stools. In the case of an overdose, offer supportive treatment and attempt to remove gastrointestinal contents. Cholestyramine has been shown to enhance the elimination of meloxicam.13
Synthesis

Figure 2. Published synthetic procedures leading to Meloxicam
Two viable synthetic pathways are described in the literature for the synthesis of meloxicam. The key intermediate of the first route (Figure 2, route A) is a 4-hydroxy-2-methyl-2H-1,2-benzothiazine-3-carboxylate 1,1-dioxide ester 3, which is reacted with 5-methyl-1,3-thiazol-2-amine (4). The second literature procedure (Figure 2, route B) for the synthesis of meloxicam (1) proceeds via 4-hydroxy-N-(5-methyl-1,3-thiazol-2-yl)-2H-1,2-benzothiazine-3-carboxamide 1,1-dioxide (5), which is methylated in position 2 in the last step, with methyl iodide,8 in the presence of a base. A common drawback of the literature procedures is the formation of impurities 6, alkylated at the nitrogen atom of the thiazole ring.14
References
1. Bekker A, Kloepping C, Collingwood S. Meloxicam in the management of post-operative pain: Narrative review. J Anaesthesiol Clin Pharmacol. 2018 Oct-Dec;34(4):450-457.
2. Moore RA, Derry S, McQuay HJ. Single dose oral meloxicam for acute postoperative pain in adults. Cochrane Database Syst Rev. 2009 Oct 7;(4):CD007552.
3. Turck D, Roth W, Busch U. A review of the clinical pharmacokinetics of meloxicam. Br J Rheumatol. 1996 Apr;35 Suppl 1:13-6.
4. Katz JA: COX-2 inhibition: what we learned--a controversial update on safety data. Pain Med. 2013 Dec;14 Suppl 1:S29-34.
5. Seddik H, Rabhi M. Meloxicam-induced colitis revealed by acute abdominal pain. Ann Pharm Fr. 2013 Mar;71(2):119-22.
6. Ricciotti E, FitzGerald GA: Prostaglandins and inflammation. Arterioscler Thromb Vasc Biol. 2011 May;31(5):986-1000.
7. Singh G, Lanes S, Triadafilopoulos G (July 2004). "Risk of serious upper gastrointestinal and cardiovascular thromboembolic complications with meloxicam".The American Journal of Medicine. 117(2): 100–106.
8."Meloxicam official FDA information, side effects, and uses". Drugs.com. March 2010. Archived from the original on 16 March 2010. Retrieved 11 Feb 2022.
9. FDA Approved Drug Products: ZYNRELEF (bupivacaine and meloxicam) extended-release solution for injection。
10. Carl P. Weiner MD, MBA, FACOG, Clifford Mason PhD (2019). Drugs for Pregnant and Lactating Women (3rd ed.). Elsevier.
11. Chesne C, Guyomard C, Guillouzo A, Schmid J, Ludwig E, Sauter T: Metabolism of Meloxicam in human liver involves cytochromes P4502C9 and 3A4. Xenobiotica. 1998 Jan;28(1):1-13.
12. FDA approved products: Mobic (meloxicam) oral tablets.
13. Medsafe NZ: Mobictab (meloxicam) oral tablets.
14. Mezei, T., Mesterházy, N., Bakó, T., Porcs-Makkay, M., Simig, G., & Volk, B. (2009). Manufacture of high-purity meloxicam via its novel potassium salt monohydrate. Organic Process Research & Development, 13(3), 567-572.
General Description
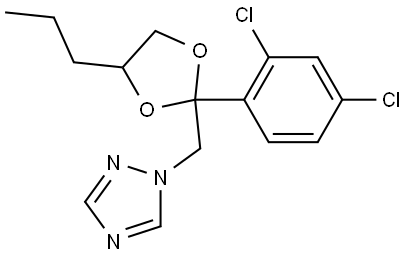
Propiconazole is the cyclic ketal obtained by formal condensation of 1-(2,4-dichlorophenyl)-2-(1H-1,2,4-triazol-1-yl)ethanone with pentane-1,2-diol. A triazole fungicide, it is used commercially as a diastereoisomeric mixture on soft fruit (including apricots, peaches, nectarines, plums and prunes), nuts (including peanuts, pecans and almonds), mushrooms, and grasses grown for seeds. It has a role as a xenobiotic, an environmental contaminant, an EC 1.14.13.70 (sterol 14alpha-demethylase) inhibitor and an antifungal agrochemical. It is a member of triazoles, a cyclic ketal, a dichlorobenzene, a conazole fungicide and a triazole fungicide.1
Propiconazole is a triazole fungicide, also known as a DMI, or demethylation inhibiting fungicide due to its binding with and inhibiting the 14-alpha demethylase enzyme from demethylating a precursor to ergosterol. Without this demethylation step, the ergosterols are not incorporated into the growing fungal cell membranes, and cellular growth is stopped. Propiconazole is used agriculturally on turfgrasses grown for seed and aesthetic or athletic value, mushrooms, corn, wild rice, peanuts, almonds, sorghum, oats, pecans, apricots, peaches, nectarines, plums and prunes. It is also used in combination with permethrin in formulations of wood preserver. Propiconazole is a mixture of four stereoisomers and was first developed in 1979 by Janssen Pharmaceutica.2

Figure 1. Properties of Propiconazole
Pharmacology and Biochemistry
Mechanism of Action
A first version cDNA microarray of the cladoceran Daphnia magna was developed. Through Suppression Subtractive Hybridization PCR (SSH-PCR) 855 life stage-specific cDNAs were collected and used to document the toxicological mode of action of the pesticide propiconazole. DNA sequencing analysis revealed gene fragments related to important functional classes such as embryo development, energy metabolism, molting and cell cycle. Major changes in transcription were observed in organisms exposed for 4 and 8 days to 1 microg/mL. After 4 days a 3-fold down-regulation of the gene encoding the yolk protein, vitellogenin, was observed indicating impaired oocyte maturation. Moreover, genes such as a larval-specific gene and chaperonin were repressed, whereas the heat shock 90 protein and ATP synthase were induced. Organismal effects clearly confirmed the major molecular findings: at the highest concentration (1 ug/mL) adult growth was significantly (p < 0.05) impaired and increased developmental effects in the offspring could be noted. We have demonstrated the potential of microarray analysis in toxicity screening with D. magna. The use of vitellogenin mRNA as a rapid biomarker of reproductive effects in chronic toxicity studies with cladocerans is suggested.3
Absorption, Distribution and Excretion
After oral administration to rats, propiconazole is rapidly absorbed and also rapidly and almost completely eliminated with urine and feces. Residues in tissues were generally low and there was no evidence for accumulation or retention of propiconazole or its metabolites. Male and female mice were fed a diet containing 5, 100 and 2500 ppm propiconazole for 21 days, followed by a single oral dose of (14C)-phenyl propiconazole at a mean corresponding dose level (0.8/1.0, 16.8/21.5 and 434/475 mg/kg bw for males/females). Urinary excretion accounted for 45-81% of the administered dose after 96 hours and tended to be higher by males than by females. In the feces 22-43% was excreted. At the lowest dose level (5 ppm propiconazole) the residual radioactivity in blood, liver, kidneys, lungs and in the remaining carcass was below 0.02 mg/kg propiconazole equivalents and accordingly higher at the 100 and 2500 ppm dose level. Residues in female mice were higher than in male mice, except in the kidneys where the males showed higher or equal values. Independent of the dose level and sex of the animals, the highest residues were found in the liver, up to 2.3 and 3.0 mg/kg in males and females of the highest dose level, respectively.4,5
Metabolism
The metabolism of propiconazole was investigated in male rats administered a single oral dose of 31.4 mg/kg triazole-(3,5-14C-propiconazole). Metabolites were isolated from first day urine and feces excretion representing 44.5% and 36.2% of the applied dose, respectively. A wide array of biotransformations occurred leading to numerous metabolites. The major site of enzymatic attack oxidation of the propyl side chain leading via alcohols and diols to carboxy acids and alpha-hydroxy carboxy acid or cleavage of the dioxolane ring. The majority of the alcoholic and phenolic metabolites are renally excreted as sulfuric acid and glucuronic acid conjugates. In the rat the main metabolite is the alpha hydroxyl carboxylic acid of propiconazole.5
Synthesis
Propiconazole (9) was synthesised from ethanone- l-(2,4-dichlorophenyl)(17) in a three stage process via l,3~dioxolane-2-(2,4-dichlorophenyl)-2-bromomethyl-4-n-propyl(18) as illustrated in Figure 2.6

Figure 2. Synthesis of Propiconazole
Ethanone-1-(2,4-dichlorophenyl)(17) was brominated to yield ethanone-2-bromol-(2,4-dichlorophenyl)(15) which then underwent a condensation reaction with 1,2-pentanediol (16) in the presence of an acid catalyst to yield 1,3-dioxolane-2-(2,4-dichlorophenyl)-2-bromomethyl-4-n-propyl (18). An alternative preparation of (18) involved the conversion of ethanone-1-(2,4-dichlorophenyl)(17) to 1,3-dioxolane-2-(2,4-dichlorophenyl)-2-methyl-4-n-propyl (182) by condensation with 1,2-pentanediol (16) followed by bromination in dichloromethane at 35°C. In the final stage l,3-dioxolane-2-(2,4-dichlorophenyl)-2-bromomethyI-4-rc-propyl (18) underwent a nucleophilic substitution reaction with the sodium or potassium salt of 1H-1,2,4-triazole (8) to yield l-[2-(2,4-dichlorophenyl)-4-propyl-1,3-dioxolan-2-ylmethyl]-1H-1,2,4-triazole (propiconazole)(9). The process outlined by Ariguard was then scaled-up to 2-litres in order to try and evaluate any problems that might occur from a future industrial scale-up. Propiconazole (9) and the intermediate products were isolated and characterised by boiling points, NMR, FT1R and GC-MS.8-11
References
1. ChEBI, http://www.ebi.ac.uk/chebi/searchId.do?chebiId=CHEBI:8489.
2. Toxin and Toxin Target Database (T3DB).
3. Soetaert A et al; Comp Biochem Physiol C Toxicol Pharmacol 142 (1-2): 66-76 (2006).
4. Tomlin CDS, ed. Propiconazole (60207-90-1). In: The e-Pesticide Manual, 13th Edition Version 3.2 (2005-06). Surrey UK, British Crop Protection Council.
5. WHO/FAO; Joint Meeting on Pesticide Residues on Propiconazole (60207-90-1). Available from, as of July 5, 2006.
6. E. Ebert and W Eckhardt, Zh. Naturforsch. 1989, 44 part.8, 85-96.
7. G. Von Reet, J Heeres and L. Wals, Janssen, Ger. Pat. 2,551-560, 1976.
8. Hadjudemetriou, D and Loetflof R., J. of Labelled Compounds and Radiopharmaceuticals 1992 31 p545-551.
9. Koltai E., Kling F. and G. Rutkai, J. of Labelled Compounds and Radiopharmaceuticals 1995 36 p903-908.
10. Agriguard Ltd., technical bulletin, November 1997.
11. R. Colle and G. Camaggi, Montedison S.A., Pat. ZA 8606825 1987.
You may like
Related articles And Qustion

See also

Lastest Price from Propiconazole manufacturers

US $0.00/kg2025-09-28
- CAS:
- 60207-90-1
- Min. Order:
- 1kg
- Purity:
- 95
- Supply Ability:
- 200000

US $0.00/KG2025-04-21
- CAS:
- 60207-90-1
- Min. Order:
- 25KG
- Purity:
- 98%min
- Supply Ability:
- 30tons/month