Convenient Preparation of Tri-tert-butylphosphine Tetrafluoroborate
Tri-tert-butylphosphine tetrafluoroborate(CAS: 131274-22-1) also called Tri-tert-butylphosphonium tetrafluoroborate (TTBP • HBF4) is used as a precursor to tri-tert-butylphosphine (TTBP), which is widely used in palladium-catalyzed cross-coupling reactions[1]. Although TTBP gives highly active catalysts for a number of reactions, it is inconvenient to use due to its low melting point (30 ◦C) and air sensitivity. Pure TTBP is pyrophoric as a solid and rapidly oxidizes in solution (< 4 min for complete oxidation of a 0.1 M solution in THF). In contrast, TTB• HBF4 is a high-melting solid that is indefinitely stable in air as a solid and in solution. TTB • HBF4 serves as an effective substitute for TTBP provided that the reaction is carried out under sufficiently basic conditions to generate the free phosphine (pKa = 11.4).Since, most Pd-catalyzed coupling reactions are carried out using a stoichiometric amount of base, TTB • HBF4 can be deprotonated in situ. A variety of bases, such as KF, CsF, Cy2NMe, HN(i-Pr)2, were shown to effectively deprotonate TTB • HBF4 in the presence of Pd2(dba)3 in THF to generate Pd(TTBP)2, which is the active catalyst precursor in Pd-catalyzed coupling reactions [2].
The introduction of basic and bulky trialkylphosphines, such as tricyclohexylphosphine and tri-tert-butylphosphine, has had a tremendous impact in transition-metal catalysis. According to the carbonyl stretching frequency of the Ni(CO)3[P(t-Bu)3] complex, tri-tert-butylphosphine is a very strong electron-donating ligand.2 It is also a sterically very bulky ligand having a large cone angle of 182°. Its electron-rich nature facilitates the oxidative addition of previously reluctant substrates (e.g., aryl chlorides) in palladium-catalyzed reactions even at ambient temperature. The steric demand of the phosphine aids the formation of coordinatively unsaturated species of crucial importance for initiating catalytic processes and its steric bulk enhances the rate of reductive elimination. Therefore, tritert-butylphosphine has found widespread application in many transition-metal-catalyzed reactions. Here, we report a procedure addressing the previous shortcomings and present a synthesis of tri-tert-butylphosphonium tetrafluoroborate from inexpensive starting materials without the need for sophisticated equipment that gives 1 reliably, in good yields, and on a 50-mmol scale.
General procedure
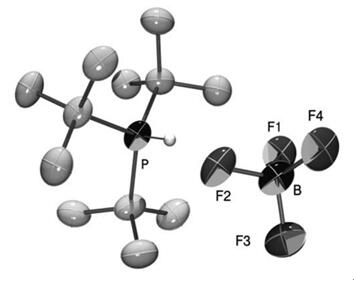
Simple extraction procedures followed by crystallization avoid any handling of air sensitive and hazardous phosphine. We use advantageously the high solubility of the tri-tert-butylphosphonium salts in aqueous solution, thus allowing the removal of uncharged and apolar byproducts by extraction with hydrocarbon or ethereal solvents. The desired phosphonium salt itself is subsequently extracted with dichloromethane. By such treatment, the obtained crude product already has of a purity of >95% (1 H NMR). The main contamination consists of di-tert-butylphosphonium tetrafluoroborate. A single recrystallization from ethanol reduces this impurity to less than 0.3% and provides 1 as crystalline material. This also allows the previously unobtained X-ray crystallographic structure of the phosphonium tetrafluoroborate to be recorded.
A synthesis of tri-tert-butylphosphonium tetrafluoroborate
To a dried 500-mL three-neck flask equipped with an addition funnel, an internal thermometer, and a magnetic stirrer bar were added CuBr•Me2S (514 mg, 2.50 mmol) and LiBr (434 mg, 5.00 mmol). The reaction vessel was purged with N2 and hexane (100 mL) was added. PCl3 (4.37 mL, 50.0 mmol) was added to the suspension and the reaction was cooled with an ice bath. 2.0 M t-BuMgCl in Et2O (100 mL, 200 mmol) was added dropwise and during the addition of the first 50 mL, the internal temperature was kept below 8 °C. The flask was warmed with an ambient water bath and the remainder of the t-BuMgCl soln was added and the mixture was stirred vigorously for 13 h at 23 °C. The mixture was then recooled with an ice bath and 3 M aq HBF4 soln (175 mL, 525 mmol) was carefully added, keeping the internal temperature below 25 °C. The biphasic mixture was stirred for 15 min and filtered over a pad of Celite. The layers were separated and the aqueous layer was washed with hexane (2 × 100 mL) to remove apolar impurities. The aqueous layer was then extracted with CH2Cl2 (3 × 200 mL). The combined CH2Cl2 layers were dried (MgSO4), filtered, and evaporated in vacuo to afford crude tri-tert-butylphosphonium tetrafluoroborate (12.0 g) as a white solid (96% NMR purity). Crystallization (EtOH, 6 mL/g) afforded analytically pure material (9.76 g) as colorless plates. The mother liquor was concentrated and crystallization (EtOH) afforded additional product (1.06 g) (75% combined yield); mp 300–302 °C (dec.) (EtOH).
- IR (ATR): 3003, 1474, 1382, 1179, 1052, 1029, 907, 885, 726 cm–1.
- 1 H NMR (400 MHz, CDCl3): d = 6.08 (d,1 JPH = 465 Hz, 1 H), 1.67 (d, 3 JPH = 15.3 Hz, 27 H). 13C NMR (100 MHz, CDCl3): d = 37.1 (d, 1 JPC = 28.8 Hz), 30.1.
- 31P NMR (162 MHz, CDCl3): d = 51.5.
- Anal. Calcd for C12H28BF4P: C, 49.68; H, 9.73. Found: C, 49.68; H, 9.63 [3].
Reference
[1]http://www.oakwoodchemical.com/ProductsList.aspx?CategoryID=-2&txtSearch=38223&ExtHyperLink=1
[2] Shaughnessy, K. H., Samaali, S. , Pinsonneault, F. and Gagnon, A. (2015). Tri‐tert‐butylphosphonium Tetrafluoroborate. In Encyclopedia of Reagents for Organic Synthesis.
[3] Saget, T. and N. Cramer (2011). "Convenient Preparation of Tri-tert-butylphosphonium Tetrafluoroborate." Synthesis 2011: 2369-2371
Related articles And Qustion



See also


Lastest Price from Tri-tert-butylphosphine tetrafluoroborate manufacturers
US $0.00/kg2025-08-13
- CAS:
- 131274-22-1
- Min. Order:
- 1kg
- Purity:
- 99%
- Supply Ability:
- 10000KGS

US $0.00-0.00/KG2025-04-21
- CAS:
- 131274-22-1
- Min. Order:
- 1KG
- Purity:
- 99%
- Supply Ability:
- 20 mt