氟红霉素的药理作用
发布日期:2020/10/24 7:56:57
背景及概述[1]
自1952年由礼来公司开发的首个大环内酯类抗生素红霉素A上市至今,60多年全球医药企业和各大研究所对该类抗生素的研究就一直没有停止过,大致可以分为三个阶段:(1)50年代~80年代的为代大环内酯类抗生素时代,有红霉素、地红霉素、麦白霉素、交沙霉素、乙酰螺旋霉素、麦迪霉素。主要用于治疗由多种革兰氏阳性菌引起的呼吸道感染等疾病,对衣原体、支原体、弯曲杆菌、军团菌等活性较强。这类化合物疗效确切、无严重的不良反应,但是对胃酸不稳定,生物利用度低,胃肠道反应较多;(2)80年代~90年代的为第二代大环内酯类抗生素时代,主要有克拉霉素、罗红霉素、阿奇霉素、罗他霉素、美欧卡霉素、氟红霉素等,这一代药物改善了代大环内酯类抗生素的酸不稳定性的缺点,提高了生物利用度,抗菌活性更强,毒性低,副作用少,延长了半衰期,是广泛用于呼吸道感染的一线药物。氟红霉素与阿奇霉素、罗红霉素等红霉素衍生物一样属于第二代红霉素类抗生素。与红霉素相比,具有抗菌作用强、范围广,在血液、组织体液及细胞内药物浓度高且持久,半衰期长,克服了红霉素在酸存在下的不稳定性、药代动力学性能改善、口服生物利用度高、不良反应明显低于红霉素,患者耐受性良好。
药理作用[1]
氟红霉素是氟化的大环内酯类抗生素,具有强的体外抗菌活性,在低的pH环境中仍然保持高度的稳定性。对于肺炎链球菌、葡萄球菌及溶血A基团链球菌等均有抑制作用,MIC为0.0015-0.006ug/ml,对流感嗜血杆菌和金黄色葡萄球菌的MIC为0.012-0.4ug/和0.1-3.1ug/ml,体内试验表明对酿脓链球菌、肺炎链球菌和金黄色葡萄球菌引起的感染的EC50值低于红霉素。本品在胃液中的t1/2时间长,组织分布率高,几乎没有副作用。本品作用机制、抗菌谱与红霉素相似,在酸性条件下稳定,口服生物利用度高,半衰期较长,体外试验显示抗菌效力与红霉素相当而略高于交沙霉素,肝细胞培养结果提示本品肝细胞毒性低于红霉素。本品对肝酶活性影响较少,对苯巴比妥的中枢抑制作用亦影响较少。
适应症[1]
上下呼吸道感染及牙和口腔感染的治疗。
其他应用[2]
氟红霉素为第二代第二代大环内酯类抗生素,但第二代大环内酯类抗生素并没有提高对大环内酯耐药菌的抑制活性,因而随着耐药菌的迅速增多,第二代大环内酯类抗生素面临新的挑战;(3)90年代至今主要发展了第三代大环内酯类抗生素,以泰利霉素为代表,其保留了对敏感菌的抗菌活性,且对、第二代大环内酯类抗生素耐药菌也有良好的活性,抗菌谱更广。氟红霉素可作为起始反应原料制备新的大环内酯类抗生素,如:
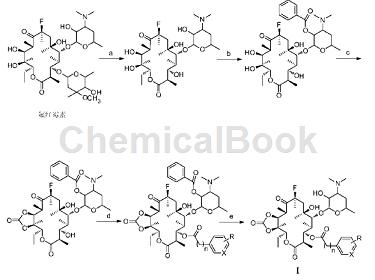
大环内酯类抗菌化合物I1的合成:
(1)将氟红霉素(70.0g,93mmol)溶于1200mLpH为1.5盐酸缓冲液中,在室温下搅拌反应24h。溶液浓缩至200mL,加氨水调节pH至9。乙酸乙酯萃取3次(150mL×3),有机相依次用饱和碳酸氢钠溶液(100mL×3),饱和食盐水溶液(100mL×3)洗涤,无水硫酸钠干燥过夜。过滤,蒸去溶剂,硅胶柱层析(展开剂V二氯甲烷/V甲醇=20:1),得白色固体产物3-脱(己吡喃葡萄糖基)-3-羟基氟红霉素(40.0g,72%)。
(2)将步骤(1)所得产物(40.0g,67.3mmol)溶于250mL无水四氢呋喃中,0℃下加入苯甲酸酐(18.5g,81.8mmol),搅拌,0.5h内缓慢滴加三乙胺9.50mL,自然升温至室温反应48h。反应液中加入饱和碳酸氢钠溶液(100mL),搅拌1h后静置,分层,留有机相。有机层依次用饱和碳酸氢钠溶液(60mL×3),饱和食盐水溶液(60mL×3)洗涤,无水硫酸钠干燥过夜。过滤,蒸去溶剂,硅胶柱层析(展开剂V二氯甲烷/V甲醇=40:1),得白色固体产物3-脱(己吡喃葡萄糖基)-3-羟基-2’-苯甲酰基氟红霉素(42.0g,89%)。
(3)将步骤(2)所得产物(42.0g,60.3mmol)溶于250mL无水二氯甲烷,0℃下加入吡啶2.1mL,缓慢滴加固体三光气(21.5g,72.4mmol)的二氯甲烷溶液(50mL),再缓慢滴加三乙胺30mL。搅拌5.5h,向反应瓶中加入少量的碎冰块终止反应。有机相依次用饱和碳酸氢钠溶液(100mL×3),饱和食盐水溶液(100mL×3)洗涤,无水硫酸钠干燥过夜。过滤,蒸去溶剂,硅胶柱层析(展开剂V石油醚/V乙酸乙酯=1:1),得淡黄色固体产物3-脱(己吡喃葡萄糖基)-3-羟基-11,12-环碳酸酯-2’-苯甲酰基氟红霉素(35.0g,80%)。
(4)将步骤(3)所得产物(1.00g,1.38mmol)溶于20mL二氯甲烷,0℃下,加入对硝基苯乙酸(0.31g,1.73mmol),EDCI(0.33g,1.73mmol),DMAP(0.017g,0.14mmol),继续搅拌0.5h后升至室温,TLC监测反应。反应毕,向反应液中加入2mol/L的氢氧化钠溶液,调节pH至8。有机相依次用饱和的NH4Cl溶液(30mL×3),饱和食盐水溶液(30mL×3)洗涤,无水硫酸钠干燥过夜。过滤,蒸去溶剂,硅胶柱层析(展开剂V石油醚/V乙酸乙酯=1:1),得黄色固体产物3-脱(己吡喃葡萄糖基)-3-对硝基苯乙酰基-11,12-环碳酸酯-2’-苯甲酰基氟红霉素。
(5)将步骤(4)所得产物(0.4g,0.45mmol)溶于10mL甲醇中,回流10h,蒸去溶剂,加入20mL乙酸乙酯,2mol/L氢氧化钠溶液调节pH至8,有机相依次用饱和的NH4Cl溶液(10mL×3),饱和食盐水溶液(10mL×3)洗涤,无水硫酸钠干燥过夜。过滤,蒸去溶剂,硅胶柱层析(展开剂V二氯甲烷/V甲醇=30:1),得黄色固体化合物3-脱(己吡喃葡萄糖基)-3-对硝基苯乙酰基-11,12-环碳酸酯-氟红霉素I1(0.2g,18%)。
主要参考资料
[1]说明书
[2]CN201510713804.0一种大环内酯类抗菌化合物及其制备方法与应用
欢迎您浏览更多关于氟红霉素的相关新闻资讯信息