Synthesis and Application of 3-O-Benzyl-4-(hydroxymethyl)-1,2-O-isopropylidene-a-D-ribofuranose
General description
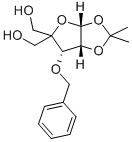
3-O-Benzyl-4-(hydroxymethyl)-1, 2-O-isopropylidene-A-D-Ribofuranose was estimated to have a boiling point of 442.5±45.0 °C and a pH coefficient of 13.95±0.10, which was required to be stored at 2-8°C. It is a useful research chemical.

Fig. 1 The structure of 3-O-Benzyl-4-(hydroxymethyl)-1,2-O-isopropylidene-a-D-ribofuranose.
Synthetic routes

Fig. 2 The synthetic step 1 of 3-O-Benzyl-4-(hydroxymethyl)-1,2-O-isopropylidene-a-D-ribofuranose.
Acetic acid in water (80%, 1L) was added to (3aR,6R,6aR)-6-(benzyloxy)-5-(2,2-dimethyl-1,3-dioxolan-4-yl)-2,2-dimethyltetrahydrofuro[2,3-d][1,3]dioxole (100 g, 0.29 mol) and the mixture stirred at r.t. for 42 h. The reaction mixture was poured into a solution of NaOH solution (540g in 3L water) with vigorous stirring then extracted with EtOAc (×3). The organic layers were combined and dried (MgSO4) then evaporated. A4 (86.7 g, 98%) as yellow oil. 1H NMR δ (300 MHz, CDCl3) δ 7.47-7.28 (m, 5H), 5.77 (d, 1H), 4.78 (d, 1H), 4.65-4.50 (m, 2H), 4.16-4.10(m, 1H), 4.03-3.97 (m, 1H), 3.92 (dd, 1H), 3.75-3.61 (m, 2H), 2.50-2.38 (m, 2H), 1.59 (s, 3H), 1.36 (s, 3H) [1].

Fig. 3 The synthetic method 2 of 3-O-Benzyl-4-(hydroxymethyl)-1,2-O-isopropylidene-a-D-ribofuranose.
A solution of A4 (23.66g, 76.2 mmol) in water (250 mL) was slowly added to a solution of sodium periodate (19.08g, 89.2 mmol) in water (125 mL) at 0°C. After 30 min., ethylene glycol (2.5 mL) was added and the reaction mixture extracted with EtOAc. The organic layers were combined, dried (MgSO4) and evaporated. A5 (20.44 g, 96%) as yellow oil. 1H NMR δ (300 MHz, CDCl3) δ 9.61 (d, 1H), 7.42-7.24 (m, 5H), 5.81 (d, 1H), 4.75 (d, 1H), 4.63(d, 1H), 4.60(t, 1H), 4.49(dd, 1H), 3.85(dd, 1H), 1.60(s, 3H), 1.36 (s, 3H) [1].

Fig. 4 The synthetic method 3 of 3-O-Benzyl-4-(hydroxymethyl)-1,2-O-isopropylidene-a-D-ribofuranose.
Aqueous 37% formaldehyde (40 mL) followed by 1N NaOH (200 mL) were added to a solution of A5 (20.44g, 73.45 mmol) in water (150 mL) and dioxane (50 mL) at 0°C. The reaction mixture was stirred at r.t. for 7 days and then partitioned between EtOAc and brine. The organic layers were combined, dried (MgSO4) and evaporated. A6 (22.79 g, 100%) as a pale yellow oil. 1H NMR δ (400 MHz, CDCl3) δ 7.41-7.28 (m, 5H), 5.76(d, 1H), 4.80 (d, 1H), 4.62 (dd, 1H), 4.52 (d, 1H), 4.21 (d, 1H), 3.90 (dd, 2H), 3.78 (dd, 1H), 3.55 (dd, 1H),2.37 (t,1H), 1.89 (dd, 1H),1.63 (s, 3H),1.33 (s, 3H) [1].
Application
Synthesis of conformationally restricted bicyclic 3 '-azido/amino-xylofuranosylpyrimidines
Conformationally rigid 3'-azido-3'-deoxy-2'-O,4'-C-methylene-xylofuranosylpyrimidines have been synthesized from dihydroxysugar precursor, 3-O-benzyl-4-C-hydroxymethyl-1,2-O-isopropylidene-a-D-ribofuranose following a convergent methodology in good yields. The dihydroxy sugar precursor was conveniently synthesized from diacetone-D-glucose following a literature procedure. As a model case, it is demonstrated that 3'-azido-3'-deoxy-2'-O,4'-C-methylene-xylofuranosylthymine can easily be converted into corresponding bicyclic 3'-amino-nucleoside in quantitative yield by its treatment with Pd-C under hydrogen atmosphere [2].
Facile synthesis and conformation of 3A-O,4A-C-methyleneribonucleosides
Bicyclic nucleoside analogues, 3'-O,4'-C-methyleneribonucleosides 1, including thymine, cytosine, adenine and guanine nucleobases, were conveniently synthesized from dglucose, and the ribofuranose ring of 1 was found to exist predominantly in a S-conformation by means of 1H NMR and X-ray analysis. the synthesis of the target compounds 1 was performed by a coupling reaction of a 1-Oacetylribofuranose derivative with silylated nucleobases and a subsequent oxetane ring formation. A stereoselective silylation of the diastereotopic hydroxy groups in 3-O-benzyl-4-hydroxymethyl-1,2-O-isopropylidene-a-dribofuranose 2 gave the desired compound 3 (67%). The stereochemistry at C4 in 3 was confirmed by means of NOE measurements. A p-tolylsulfonylation of 3 afforded the tosylate 4 (97%), which was converted to diacetate 5 (86%) by treatment with AcOH and Ac2O in the presence of a catalytic amount of H2SO4. Debenzylation and subsequant acetylation of the 3-hydroxy group in 5 gave the triacetate 6 (91%). The reaction of 6 with O,O'-bis(trimethylsilyl)thymine (T·2TMS) under Vorbrüggen’s conditions afforded only the β-anomer of thymidine derivative 7a (77%). The triacetate 6 was also coupled with silylated N4-benzoylcytosine (CBz·2TMS), N6- benzoyladenine (ABz·2TMS) and N2-isobutyrylguanine (GiBu·3TMS) to give the corresponding β-nucleoside derivatives 7b (82%), 7c (70%) and 7d (72%), respectively. Methanolysis of 7 gave diols 8 (63–90%) and then oxetane ring formation from 8 was accomplished on treatment with sodium hexamethyldisilazide in THF at room temperature, yielding only the corresponding 3'-O,4'-C-methyleneribonucleoside derivatives 9 (78–100%). The desired products 1 were obtained (65–71%) by removal of a TBDPS group in 9. We have, thus, achieved a facile synthesis of 3'-O,4'-C-methyleneribonucleosides 1 in good yield [3].
References
[1] Mayes B A, Stewart A J, Moussa A M. Preparation of D-amino acid compounds, particularly D-acids linked to therapeutic nucleoside analogs, for use in the treatment of liver diseases[P]. PCT Int. Appl., 2013177219, 2013.
[2] Nguyen H, Pan Y, Al Nahain A, et al. Synthesis of Cholesterol Glycoconjugates for Targeted Liposomal Drug Delivery[J]. Trends in Carbohydrate Research, 2020, 12(3): 1-9.
[3] Obika S, Morio K, Hari Y, et al. Facile synthesis and conformation of 3′-O, 4′-C-methyleneribonucleosides[J]. Chemical Communications, 1999 (23): 2423-2424.
You may like
See also
Lastest Price from 3-O-Benzyl-4-(hydroxymethyl-1,2-O-isopropylidene)-alpha-D-erythropentofuranose manufacturers

US $8.80-2.20/kg2025-06-28
- CAS:
- 63593-03-3
- Min. Order:
- 1kg
- Purity:
- 99%
- Supply Ability:
- 100kg

US $0.00-0.00/g2025-04-21
- CAS:
- 63593-03-3
- Min. Order:
- 50g
- Purity:
- 98%min
- Supply Ability:
- 20kg/month