Sitagliptin phosphate: Mechanism,Pharmacokinetics and Pharmacodynamics
Introduction
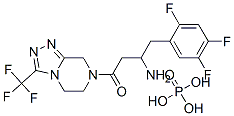
Sitagliptin phosphate (Figure 1) is chemically described as 7-[(3R)-3-amino-1-oxo-4-(2,4,5-trifluorophenyl)butyl]-5,6,7,8-tetrahydro-3-(trifluoromethyl)-1,2,4-triazolo[4,3-a]pyrazine phosphate (1:1) monohydrate. The compound has a molecular weight of 523.32 Da and amolecular formula of C16H15F6N5O·H3PO4·H2O.[1] Sitagliptin phosphate is the most commonly prescribed gliptin to treat diabetes mellitus, alone or with metformin. Sitagliptin phosphate binds to the dipeptidyl peptidase 4 enzyme, inhibiting the breakdown of glucagonlike peptide (GLP)-1 and glucose-dependent insulinotropic peptide (GIP). These hormones are released by the gut after meals and target the pancreas by raising glucose-dependent insulin secretion and decreasing glucagon output from pancreatic α-cells. Sitagliptin phosphate has many side effects including arthralgia, myopathy, pruritus, pancreatitis and it may increase hepatic enzymes level.[2]

Mechanism of Action
Sitagliptin enhances the effects of the incretin hormones glucose-dependent insulinotropic peptide (also known as gastric inhibitory polypeptide [GIP]) and GLP-1. Secreted in the intestine in response to food, GIP and GLP-1 have a role in the regulation of glucose homeostasis. Activation of GIP and GLP-1 receptors on pancreatic β-cells leads to increased levels of cyclic adenosine monophosphate and intracellular calcium, with subsequent glucose-dependent insulin secretion. In addition, sustained receptor activation is associated with insulin biosynthesis and stimulation of β-cell proliferation. Animal and in vitro data further suggest that activation of GIP and GLP-1 receptors promotes β-cell resistance to apoptosis, proliferation, and neogenesis, resulting in enhanced β-cellfunction. Additional functions of GLP-1 include inhibition of glucagon secretion from pancreatic α-cells,resulting in decreased hepatic glucose production;slowing of gastric emptying; suppression of food intake; and enhancement of glucose disposal via neuralmechanisms.
Two viable methods to enhance GLP-1 effects in vivo include administration of agents that mimic the effects of the incretins but are resistant to degradation by DPP-4 (eg, exenatide) and agents that prevent incretin degradation. Sitagliptin exerts its therapeutic effect via the latter mechanism. Thus, following administration of sitagliptin, postprandial levels of active GLP-1 are increased and activity is prolonged,with a resultant rise in insulin release and decrease inglucagon secretion from the pancreatic α-cells.[1]
Pharmacokinetics
Key pharmacokinetic parameters in healthy subjects, as provided by the manufacturer, are summarized in Table 1. Following administration of an oral 100-mg dose in healthy volunteers, sitagliptin phosphate was rapidly absorbed,with a median Tmax of 1 to 4 hours. Plasma AUC was 8.52 µM·h and Cmax was 950nM. Sitagliptin phosphate plasma AUC has been found to be increased in an approximate dose-dependent manner in both single-dose(1.5-600 mg) and multiple-dose (25-600 mg QD and 300 mg BID) studies in healthy volunteers,whereas Cmax increased in a slightly greater than dose-proportional manner. The administration of a high-fat breakfast prior to a single oral 25-mg dose of sitagliptin phosphate has not been found to influence the plasma AUC0-∞; the ratio of the least-squares (LS) mean (95% CI) ratio (fed/fasted) was 1.01 (0.94-1.10). An increase in Cmax of ~20% was observed in the fed state; however, this difference was not statistically significant versus that observed in the fasting state (LS mean ratio, 1.21 [95% CI, 1.00-1.45]).Since no pharmacokinetic parameters are appreciably influenced by food, sitagliptin may be dosed without regard to meals. Steady-state concentrations of sitagliptin are achieved within 2 to 3 days of administration. The mean volume of distribution of sitagliptin, as determined after administration of a single 100-mg IV dose in healthy subjects, is ~198 L, and 38% of the drug is reversibly bound to plasma proteins.[1]

Metabolism
Sitagliptin phosphate is mostly not metabolised, with 79% of the dose excreted in the urine as the unchanged parent compound. Minor metabolic pathways are mediated mainly by cytochrome p450(CYP)3A4 and to a lesser extent by CYP2C8. After 18 hours, 81% of the dose has remained unchanged, while 2% has been N-sulfated to the M1 metabolite, 6% has been oxidatively desaturated and cyclized to the M2 metabolite, <1% glucuronidated at an unknown site to the M3 metabolite, <1% has been carbamoylated and glucuronidated to the M4 metabolite, 6% has been oxidatively saturated and cyclized to the M5 metabolite, and 2% has been hydroxylated at an unknown site to the M6 metabolite. The M2 metabolite is the cis isomer while the M5 metabolite is the trans isomer of the same metabolite.[3]
Route of elimination
Approximately 79% of sitagliptin phosphate is excreted in the urine as the unchanged parent compoundLabel. 87% of the dose is eliminated in the urine and 13% in the feces[4]
Pharmacodynamics
The pharmacodynamic effects of sitagliptin phosphate in patients with type 2 diabetes have been described in a randomized, double-blind, placebo-controlled, 3-period,single-dose, crossover study involving 56 patients. After an overnight fast, patients received, in randomized order, sitagliptin 25 or 200 mg and placebo, with a 7-day washout period in between treatments. As in healthy subjects, in patients with type 2 diabetes,treatment with sitagliptin phosphate was associated with dose dependent inhibition of plasma DPP-4 activity. The mean (95% CI) percentages of inhibition of plasma DPP-4 activity over a 24-hour period with the 25- and 200-mg doses and placebo were 68.1% (66.6% to69.6%), 91.4% (90.9% to 91.8%), and 2.1% (-2.8% to 6.7%), respectively; the difference between either sitagliptin phosphate dose and placebo was statistically significant (P < 0.001). The difference between sitagliptin phosphate doses was also significant (P< 0.05). After an oral glucose tolerance test (OGTT) administered 2 hours postdose, the weighted average augmentation (WAA) active GLP-1 and GIP levels in either sitagliptin dose were both ~2-fold greater than those observed with placebo (P<0.001). Higher levels of WAA active GLP-1 (1.3- and 1.9-fold for the 25- and 200-mg doses,respectively) and WAA active GIP (1.4- and 2-fold for the 25- and 200-mg doses, respectively) compared with placebo were also observed following an OGTT given24 hours postdose (P < 0.001). The differences between the 25-and 200-mg sitagliptin phosphate doses were also significant for both the WAA active GLP-1 and WAA active GIP levels (P < 0.001).[5]
References
[1]Zerilli T, Pyon EY. Sitagliptin phosphate: a DPP-4 inhibitor for the treatment of type 2 diabetes mellitus. Clin Ther. 2007;29(12):2614-2634. doi:10.1016/j.clinthera.2007.12.034
[2] Zaid AN, Abu Zaaror Y, Kaddumi A, et al. Stability of extemporaneously prepared sitagliptin phosphate solution. PLoS One. 2022;17(3):e0262068. Published 2022 Mar 16. doi:10.1371/journal.pone.0262068
[3] Vincent SH, Reed JR, Bergman AJ, et al. Metabolism and excretion of the dipeptidyl peptidase 4 inhibitor [14C]sitagliptin in humans. Drug Metab Dispos. 2007;35(4):533-538. doi:10.1124/dmd.106.013136
[4] Richter B, Bandeira-Echtler E, Bergerhoff K, Lerch C. Emerging role of dipeptidyl peptidase-4 inhibitors in the management of type 2 diabetes. Vasc Health Risk Manag. 2008;4(4):753-768.
[5] Bergman AJ, Stevens C, Zhou Y, etal. Pharmacokinetic and pharmacodynamic properties of multipleoral doses of sitagliptin, a dipeptidyl peptidase-IV inhibitor: Adouble-blind, randomized, placebocontrolled study in healthy malevolunteers. Clin Ther. 2006;28:55-72.


Lastest Price from Sitagliptin phosphate manufacturers

US $0.00/Kg/Bag2025-04-21
- CAS:
- 654671-78-0
- Min. Order:
- 2Kg/Bag
- Purity:
- 99% up, High Density
- Supply Ability:
- 20 tons

US $150.00/kg2025-04-21
- CAS:
- 654671-78-0
- Min. Order:
- 1kg
- Purity:
- 99%
- Supply Ability:
- 500kg