Epirubicin hydrochloride: Pharmacodynamic,Pharmacokinetic Properties and its application
Introduction
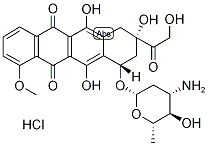
The anthracycline family drug epirubicin hydrochloride (EPR, Figure 1) is widely used in the therapy of lymphoma,leukemia, and sarcoma. These drugs localize to the nucleus of the cancer cells by forming a complex with DNA through the intercalation mode of binding. This intercalation inhibits the activity of Topoisomerase II, which further inhibits DNA replication, thereby interfering with RNA and protein synthesis. Epirubicin hydrochloride, the 4’-epimer of Doxorubicin, displayed a similar tumor response during clinical trials of its application to breast cancer and the therapeutic effects. Hence, these properties of epirubicin hydrochloride are important as they have direct consequences in clinical research.[1]

Pharmacodynamic Properties[2]
Epirubicin is the epimer of doxorubicin, with inversion of the 4'-hydroxyl group on the sugarmoiety. Epirubicin hydrochloride is a cell cycle phase non-specific anthracycline, with maximal cytotoxic effects in the Sand G2 phases. In vitro studies showed that epirubicin possesses cytotoxicity at least equivalent to that of doxorubicin against a variety of animal and human tumour cell lines including those derived from breast, liver, lung, gastric, colorectal, squamous cell, cervical, bladder,ovarian carcinomas, neuroblastoma and leukaemia. In an in vitro murine model, epirubicin hydrochloride was at least as effective as doxorubicin in inhibiting basement membrane degradation, a property deemed necessary to prevent development of metastases. Significant correlations have been detected between the in vitro activity of epirubicin hydrochloride and other anthracyclines against various tumour specimens, and therapeutic response. Multidrug resistance to anthracyclines, vinca alkaloids, dactinomycin and epidopodophyllotoxins has been reported in a variety of tumour cell lines. In vivo antitumour activity of epirubicin hydrochloride was also comparable with that of doxorubicin against a variety of human tumour xenografts in mice, including breast, lung, ovarian, prostate and testicular neoplasms.
The mechanism of anti tumour action for epirubicin hydrochloride has not been completely elucidated; however, anthracyclines appear to form a complex with DNA by intercalation between the DNA strands, thus inhibiting replication and transcription. This action may be attributed, at least in part, to interference with topoisomerase-DNA 'cleavable complex' and helicase activity by anthracyclines. Reduction of anthracyclines to semiquinone free radicals may cause damage to DNA,cell membrane lipids and mitochondria.
Myelotoxicity, which seems to be mediated, at least in part, via chromatid breaks in bone marrow cells, was lower with epirubicin hydrochloride than doxorubicin in pharmacodynamic studies. Anthracyclines appear to exert their cardiotoxic effects through a variety of mechanisms including impairment of heart mitochondrial function, depressed adenosine diphosphate-stimulated respiration, and changes in membrane structure and function. In a number of acute and chronic studies which measured these effects objectively in animals, epirubicin demonstrated a significantly lower propensity to produce cardiotoxic effects than doxorubicin, possibly due to a more rapid myocardial release of epirubicin hydrochloride during the post-infusion period.
Pharmacokinetic Properties
Following rapid intravenous administration, epirubicin hydrochloride undergoes triphasic plasma elimination. Epirubicin exhibits a rapid initial (ex) distribution phase (t1/2α = 1.8 to 4.8 minutes), followed by an intermediate (β) phase (t1/2β = 0.5 to 2.6 hours) and a much slower (γ) terminal eliminationphase (t1/2γ= 15 to 45 hours). The t1/2γ of doxorubicin was approximately 40 to 70% longer than that of epirubicin hydrochloride in the majority of comparative pharmacokinetic studies in cancer patients.However, peak plasma drug concentrations were similar following intravenous administration ofequimolar doses. Epirubicin hydrochloride undergoes extensive tissue distribution; volume of distribution values were high and variable (13 to 52L/kg), but similar to those reported for doxorubicin. Area under the plasma concentration versus time curve values adjusted for dose were 30 to 70% higher for doxorubicin than epirubicin hydrochloride following single-dose intravenous administration. Following intravenous administration, epirubicin hydrochloride is rapidly metabolised to 2 glucuronides, plus epirubicinol and 4 aglycones. Epirubicin hydrochloride is eliminated primarily via the hepatobiliary system, with approximately II to 15%of a dose eliminated in the urine as unchanged drug and metabolites. Patients with moderate to severe hepatic dysfunction exhibited reduced clearance of epirubicin hydrochloride and elevated plasma drug concentrations.[2]
Thermal Reversibility and Structural Stability in Lysozyme Induced by Epirubicin Hydrochloride
Herein Khamari et al. report the binding interactions between lysozyme (Lyz) and an anthracycline drug, epirubicin hydrochloride (EPR), through an extensive spectroscopic approach at both ensemble average and single molecular resolution. Our steady-state and time-resolved fluorescence spectroscopy reveals that the drug-induced fluorescence quenching of the protein proceeds through a static quenching mechanism. Isothermal titration calorimetry (ITC) and steady-state experiments reveal almost similar thermodynamic signatures of the drug-protein interactions. The underlying force that plays pivotal roles in the said interaction is hydrophobic in nature, which is enhanced in the presence of a strong electrolyte (NaCl). Circular dichroism (CD) spectra indicate that there is a marginal increase in the secondary structure of the native protein (α-helical content increases from 26.9 to 31.4% in the presence of 100 μM epirubicin hydrochloride) upon binding with the drug. Fluorescence correlation spectroscopy (FCS) was used to monitor the changes in structure and conformational dynamics of Lyz upon interaction with EPR. The individual association (Kass=0.33×106ms-1 M-1) and dissociation (Kdiss=1.79ms-1) rate constants and the binding constant (Kb=1.84×105 M-1) values, obtained from fluctuations of fluorescence intensity of the epirubicin hydrochloride-bound protein, have also been estimated. AutoDock results demonstrate that the drug molecule is encapsulated within the hydrophobic pocket of the protein (in close proximity to both Trp62 and Trp108) and resides ∼20 Å apart from the covalently labelled CPM dye. Förster resonance energy transfer (FRET) studies proved that the distance between the donor (CPM) and the acceptor (epirubicin hydrochloride) is ~22 Å, which is very similar to that obtained from molecular docking analysis (~20 Å). The system also shows temperature-dependent reversible FRET, which may be used as a thermal sensor for the temperature-sensitive biological systems.[1]
In vitro compatibility and stability of admixtures containing epirubicin hydrochloride
The etoposide, doxorubicin hydrochloride, vincristine sulphate, cyclophosphamide and prednisone (EPOCH) chemotherapy regimen is effective in patients with relapsed or refractory non-Hodgkin's lymphoma. However, vincristine and doxorubicin hydrochloride are relatively toxic, leading to neurovirulence and cardiotoxicity, respectively. In this study, we replaced these drugs with vindesine and epirubicin hydrochloride to reduce the cardiotoxicity and evaluated admixtures containing these drugs along with etoposide in a single infusion bag in vitro. The appearance and pH of the admixtures were evaluated, and the number of particles was detected. High-performance liquid chromatography was used to measure the concentration and degradation rates of etoposide, epirubicin hydrochloride and vindesine sulphate in each admixture. No precipitation occurred when mixing clinically relevant concentrations of etoposide, epirubicin hydrochloride and vindesine sulphate in a 0.9% NaCl injection solution. Furthermore, the delta pH of the admixtures was ≤0.12 throughout the experiment, and the number of particles (≥10 and ≥25 μm) in the solutions over the 24 hours post-preparation period met USP standards. Etoposide, epirubicin hydrochloride and vindesine sulphate were retained at >96% of their initial concentrations in the admixtures at 25°C over the course of the experiment. Etoposide, epirubicin hydrochloride and vindesine sulphate are compatible when mixed in a 0.9% NaCl injection solution, and the admixtures are stable for at least 24 hours when stored in infusion bags. This in vitro analysis indicates the suitability of our novel admixtures containing less toxic drug equivalents in a single infusion bag for clinical application.[3]
References
[1] Khamari L, Pramanik U, Shekhar S, Mohanakumar S, Mukherjee S. Thermal Reversibility and Structural Stability in Lysozyme Induced by Epirubicin Hydrochloride. Langmuir. 2021;37(11):3456-3466. doi:10.1021/acs.langmuir.1c00179
[2]Plosker GL, Faulds D. Epirubicin. A review of its pharmacodynamic and pharmacokinetic properties, and therapeutic use in cancer chemotherapy. Drugs. 1993;45(5):788-856. doi:10.2165/00003495-199345050-00011
[3]Li J, Yao C, Xu Y, Ping P, Yin H, Sun Y. In vitro compatibility and stability of admixtures containing etoposide, epirubicin hydrochloride and vindesine sulphate in a single infusion bag. J Clin Pharm Ther. 2019;44(6):875-882. doi:10.1111/jcpt.13007
You may like
See also

Lastest Price from Epirubicin hydrochloride manufacturers

US $0.00/g/Bag2025-04-21
- CAS:
- 56390-09-1
- Min. Order:
- 1g
- Purity:
- 98%min
- Supply Ability:
- 1000g

US $1.00/kg2025-04-21
- CAS:
- 56390-09-1
- Min. Order:
- 1kg
- Purity:
- 99%
- Supply Ability:
- 10 mt