Afatinib (BIBW 2992) is the second-generation potent and irreversible dual inhibitor of the epidermal growth factor receptor (EGFR) and human epidermal growth factor receptor 2 (HER2) tyrosine kinase, developed by Boehringer Ingelheim, Germany. It is capable of irreversibly inhibiting the activity of the tyrosine kinase by undergoing the Michael reaction with the thiol group of cysteine at position 797 of the EGFR. On July 12, 2013, it became a new drug for anti-small cell lung cancer approved by the US FDA under the trade name of Gilotrif. This drug is a tablet. It is used for the treatment of the patients diagnosed with metastatic non-small cell lung cancer (NSCLC) with the loss of the 19th exon or L858R mutation in the 21th exon of the tumor epidermal growth factor receptor (EGFR) confirmed using the kit approved by FDA. The drug is also effective in the treatment of HER2-positive patients with advanced breast cancer.
Boehringer Ingelheim Pharmaceuticals has applied for the protection of this compound by applying patents JP2004516283 US2010010023 WO2002050043 and so on.
Lung cancer is the world's top tumor killer with the incidence in male being higher than that in women. The number of annually new diagnosed lung cancer patients can reach 1.6 million. However, lung cancer is not just a disease. Studies have shown that lung cancer contains many different types with different types of lung cancer needs to be given specific treatment. A specific subtype of lung cancer can be determined by EGFR (ErbB receptor family members) mutation detection. These patients are also those who can benefit from the most during the treatment with Afatinib in clinical trials.
The registration approval for Afatinib in the United States is based on data from the pivotal LUX-Lung 3 trial. This trial had conducted comparison study on the Afatinib versus pemetrexed/cisplatin proposal. Data from the LUX-Lung 3 trial have demonstrated that patients who have received first-line therapy of Afatinib achieved a survival time of one year (median progression-free survival (PFS) period is about 11.1 months) before tumor growth resumed, While the value for patients receiving pemetrexed/cisplatin was slightly more than six months (PFS 6.9 months). In addition, NSCLC patients with two of the most common EGFR mutations (Del19 or L858R) had experienced a progression-free survival of over one year after receiving afatinib treatment (PFS, 13.6 months), whereas patients in the control group got a PFS of slightly more than six months (PFS 6.9 months).
In addition, compared with patients receiving standard chemotherapy, the lung cancer symptoms and quality of life of patients receiving afatinib have also been greatly improved.
In the afatinib treatment group, the most common three classes of adverse events associated with drugs were diarrhea (14%), rash (16%) and thyroid inflammation (paronychia) (11%). In the chemotherapy (pemetrexed/cisplatin), the most common drug-related AE was neutropenia (15%) and neutropenia (13%) and leukopenia (8%). Low incidence of discontinuation associated with treatment-related adverse events had been observed in the trial (8% discontinuation in the afatinib group and 12% discontinuation in the chemotherapy group). In the afatinib-treated group, only 1% of patients have discontinued the medication because of drug-related diarrhea.
This information is compiled and edited by Xiao Nan of chemicalbook.
The ability of the tyrosine kinase domains of the EGFR and HER2 to modify other proteins is critical to their ability to signal cells to grow and divide.
Afatinib is an irreversible kinase inhibitor and binds to the kinase domains of EGFR (ErbB1), HER2 (ErbB2), and HER4 (ErbB4) to inhibit tyrosine kinase autophosphorylation. This results in a downregulation of ErbB signalling and subsequent inhibition of proliferation of cell lines expressing wild-type EGFR, selected EGFR exon 19 deletion mutations, or exon 21 L858R mutations. It also inhibited in vitro proliferation of cell lines overexpressing HER2. Overall, tumour growth was inhibited.
References
http://www.fda.gov/Drugs/InformationOnDrugs/ApprovedDrugs/ucm360574.htm https://www.drugbank.ca/drugs/DB08916
Afatinib belongs to a class of drugs known as tyrosine kinase inhibitors. Tyrosine kinase inhibitors are designed to block the action of a specific enzyme called tyrosine kinase. This enzyme plays a big role in the function of cells, and is active in promoting tumor growth and progression. Afatinib works to inhibit the function of two types of tyrosine kinases: epidermal growth factor receptor (EGFR) and Her2, which are "over-expressed" by several types of cancer. By blocking the function of these tyrosine kinases, afatinib may prevent cancer cells from dividing and growing.
Afatinib was approved by the United States Food and Drug Administration (FDA) as the first-line treatment for a subset of patients with advanced non-small cell lung cancer. The patients for which afatinib was approved have lung cancer that carries a particular kind of mutation that results in an abnormal EGFR protein. These patients are most likely to be of Asian descent, women, and never smokers with a form of lung cancer known as bronchoalveolar adenocarcinoma. Clinical experience with erlotinib and gefitinib has shown that these patients respond particularly well to tyrosine kinase inhibitors that block the EGFR. This proved to be true for afatinib as well, which increased the progression-free survival of this group of patients to 13.6 months as compared to only 6.9 months for patients treated with standard chemotherapy. Erlotinib is also approved in the United States as a first-line treatment for this subgroup of patients. Longer clinical experience will be needed to determine which drug is the best choice. Afatinib may be somewhat more potent than erlotinib, but its side effects, primarily rash and diarrhea, are also more severe.
The afatinib (BIBW2992) can irreversibly inhibit the EGFR/HER2, including EGFR (wt), EGFR (L858R), EGFR (L858R/T790M) and HER2. In cell-free system, the IC50 was 0.5 nM, 0.4 nM, 10 nM and 14 nM, respectively; it has over 100-fold activity against the Gefitinib-resistant L858R-T790M EGFR mutant.
BIBW2992 is capable of irreversibly inhibiting the EGFR/HER2 with the IC50 values of wild-type EGFR, L858R mutant EGFR, L858R/T790M double mutant EGFR and HER2 being respectively 0.5, 0.4, 10 and 14 nM. For in vitro studies, BIBW2992 exhibited potent activity regardless of its effect on either wild type EGFR/HER2 or mutant EGFR/HER2, which was similar to the activity of gefitinib's effect on L858R mutant EGFR. However, the activity is 100 times as strong as the activity of gefitinib’s effect on the L858R-T790M double mutation Type EGFR. In vitro, BIBW2992 can effectively inhibit the EGFR/HER phosphorylation of all experimental types of cells.
For example, wild-type EGFR expressed in human epithelial cell carcinoma A431 cells, wild-type HER2 transfected with murine NIH-3T3 cells, and endogenous HER2 expressed in breast cancer BT-474 cell line and gastric cancer NCI-N87 cell line.
In vivo study, according to the experimental weight, oral administration of 20 mg per kilogram of BIBW2992 can last for 25 days, causing significant decline of the tumor. Through the immunohistochemical staining of the tumor biopsy, it has been found that EGFR and AKT phosphorylation have been inhibited. Thus, similar as lapatinib and neratinib, BIBW2992 is a secondary generation tyrosine kinase inhibitor being capable of irreversibly inhibiting human EGFR2 (HER2) and EGFR kinase. BIBW2992 can not only effectively inhibit the EGFR mutation, like the first-generation tyrosine kinase drugs, erlotinib and gefitinib, but also effective in dealing with EGFR mutants that are resistant to erlotinib and gefinib.
In July 2013, the US FDA approved afatinib (also referred to as BIBW- 2992), for the first-line treatment of patients with metastatic non-small cell lung cancer (NSCLC) and with tumors that have epidermal growth factor receptor (EGFR) exon 19 deletions or exon 21 (L858R) substitution. Afatinib functions as an irreversible inhibitor by covalently binding directly to the ATP-binding site in the kinase domains of both EGFR(Cys 773) andHER2 (Cys 805;HER-2 is the preferred dimerization partner of EGFR) resulting in downregulation of EGFR signaling. Afatinib is a potent inhibitor of wild-type and mutant forms (L858R) of EGFR (IC
50s of 0.5 and 0.4 nM, respectively), and HER2 (IC
50=14 nM), but about 100-fold more active against the gefitinib resistant L858R– T790M EGFR double mutant, with an IC
50 of 10 nM. Consistent with its in vitro activity, afatinib induces tumor regression in xenograft and transgenic lung cancer models, with superior activity over erlotinib. A synthetic route to afatinib that employs the displacement of a phenylsulfonyl group to install the (S)-3-hydoxytetrahydrofuran ring and a modified Horner–Wadsworth–Emmons reaction with {[4-(3-chloro-4- phenylamino)-7-((S)-tetrahydrofuran-3-yloxy)-quinazolin-6-ylcarbamoyl]- methyl}-phosphonate and dimethylaminoacetaldehyde-hydrogen sulfite adduct to install the eneamide moiety, has been reported.

Boehringer-Ingelheim (United States)
BIBW2992 (Afatinib, Tomtovok, Tovok) irreversibly inhibits EGFR/HER2 including EGFRwt, EGFRL858R, EGFRL858R/T790M and HER2 with IC50 of 0.5 nM, 0.4 nM, 10 nM and 14 nM, respectively.
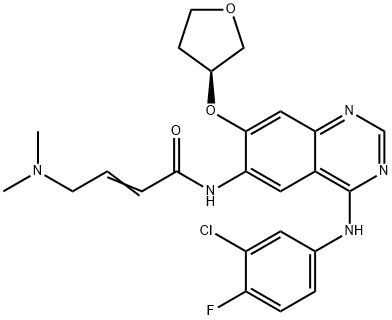
ChEBI: Afatinib is a quinazoline compound having a 3-chloro-4-fluoroanilino group at the 4-position, a 4-dimethylamino-trans-but-2-enamido group at the 6-position, and an (S)-tetrahydrofuran-3-yloxy group at the 7-position. Used (as its dimaleate salt) for the first-line treatment of patients with metastatic non-small cell lung cancer. It has a role as a tyrosine kinase inhibitor and an antineoplastic agent. It is a member of quinazolines, a member of furans, an organofluorine compound, an enamide, an aromatic ether, a tertiary amino compound, a member of monochlorobenzenes and a secondary carboxamide.