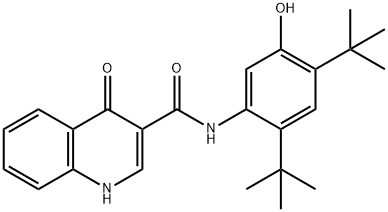
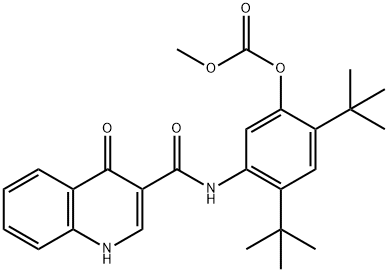
Description
In January 2012, the US FDA approved ivacaftor for the treatment of cystic
fibrosis (CF) in patients who have the G551D mutation of the CF transmembrane regulator (CFTR) and are at least 6 years old. Ivacaftor (also known as VX-770) is a CFTR potentiator that increases the open probability of CFTR, thus increasing chloride
secretion particularly in the 5% of CF patients with the G551D/F508 gating/
processing mutation. Ivacaftor was discovered by medicinal chemistry optimization of a lead scaffold identified through high-throughput screening of a 228,000 compound collection. In cultured bronchial epithelial cells from a CF patient with F508del, ivacaftor increased chloride secretion
(EC
50=81 nM). Preparation of ivacaftor is accomplished via a multistep
synthesis oftwointermediates, 4-oxo-1,4-dihydroquinoline-3-carboxylic acid
and 5-amino-di-tert-butylphenyl methyl carbonate, which are coupled using
propane phosphonic acid anhydride (T3P) to afford the amide; deprotection of
the phenol then provides ivacaftor.
Originator
Vertex Pharmaceuticals (United States)
Uses
Ivacaftor (VX-770, Kalydeco) is a potentiator of CFTR targeting G551D-CFTR and F508del-CFTR with EC50 of 100 nM and 25 nM, respectively
Uses
Ivacaftor is used in the treatment of cystic fibrosis.
Definition
ChEBI: An aromatic amide obtained by formal condensation of the carboxy group of 4-oxo-1,4-dihydroquinoline-3-carboxylic acid with the amino group of 5-amino-2,4-di-tert-butylphenol. Used for the treatment of cystic fibrosis.
Mechanism of action
Ivacaftor is an oral agent that increases the ion-function of activated cell-surface CFTR. In vitro studies using bronchial epithelial cells from the lungs of patients with CF have demonstrated that increases in the air-surface fluid level and ciliary beat frequency can be obtained by correcting abnormal CFTR-mediated ion transport. Possible improvement in airway obstruction could occur by decreasing mucus plugging through better hydration of the airway surface and increased mucus clearance. Ivacaftor potentiates CFTR to restore chloride gating function in CFTR with class III gating defects, such as with the G551D variant[3-4].
Pharmacokinetics
Administered as an oral dose, ivacaftor absorbs readily from the gut but has low solubility in water (< 0.05 ug/mL). Taking a 150 mg dose of ivacaftor with a high-fat meal improves absorption, increases AUC by 2.5 times, and delays Tmax from 3 to 5 hours. Dose/time pharmacokinetics follows a linear profile to a dose of 250 mg, though Cmax plateaus for doses 375 mg and higher. Ivacaftor is transported in the plasma highly bound (99%), preferentially to alpha-1-acid glycoprotein and albumin, to its site of action, the apical membrane of epithelial cells. However, no drug-drug interactions related to protein binding competition are expected.
Clinical Use
Vertex’s ivacaftor was granted breakthrough therapy designation by the FDA in January 2012 for
cystic fibrosis (CF) patients who bear the G551D mutation in the Cycstic Fibrosis Transmembrane
Regulator (CFTR) gene. This CFTR mutation occurs in roughly 4% of the 30,000 people living with
CF in the United States. While the compound has been identified as a potentiator in cell-based assays,
its mechanism of action is as yet unknown.
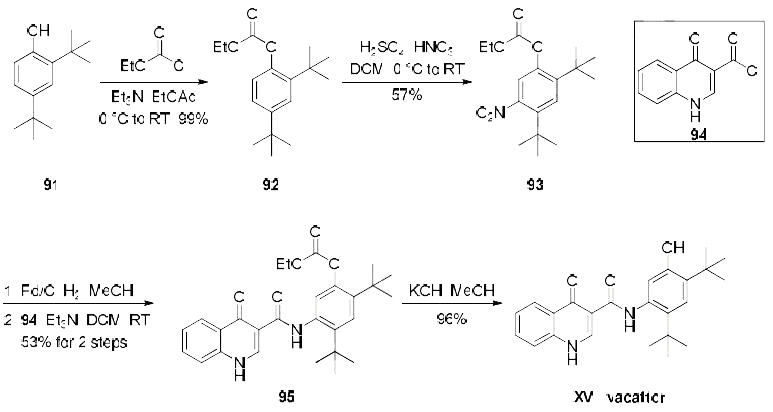
Synthesis
Several patents describe a synthesis of
ivacaftor, only one demonstrates the synthesis on scale and includes yields, which is depicted in the
scheme. Beginning with treatment of commercial di-tert-butylphenol derivative 91 with ethyl chloroformate,
the synthesis of carbonate 92 was achieved in quantitative yield. Nitration of 92 provided the desired nitroarene regioisomer 93 in 57% yield which was isolated by recrystallization. Reduction of the newlyinstalled
nitro group and subsequent amide bond formation via reaction with commercially available
acid chloride 94 produced amide 95 in 53% yield over the two step sequence. Finally, cleavage of the
carbonate unmasked the phenol to furnish ivacaftor (XV) in 96% yield.
Metabolism
Ivacaftor is metabolized in the liver by cytochrome P450 3A (CYP3A), including both CYP3A4 and CYP3A5, into a metabolite hydroxymethyl-ivacaftor (M1), which is considered to be active with a potency 1/6th that of ivacaftor itself, and the inactive metabolite ivacaftor-carboxylate (M6), which not considered active and has an activity level 1/50th that of ivacaftor. The parent drug and metabolites are eliminated predominantly (87%) through the bile, with 22% being the M1 metabolite and 43% being the M6 metabolite. The M6 metabolite is excreted in the bile through the solute carrier organic anion transporter 1B1 (SLCO1B1) transporter, but the mechanism for eliminating M1 is unknown. Ivacaftor has a half-life of 12–14 hours[3].
References
[1] van goor f1, hadida s, grootenhuis pd, burton b, cao d, neuberger t, turnbull a, singh a, joubran j, hazlewood a, zhou j, mccartney j,arumugam v, decker c, yang j, young c, olson er, wine jj, frizzell ra, ashlock m, negulescu p. rescue of cf airway epithelial cell function in vitro by a cftr potentiator, vx-770. proc natl acad sci u s a. 2009 nov 3;106(44):18825-30.
[2] vachel l1, norez c, becq f, vandebrouck c. effect of vx-770 (ivacaftor) and oag on ca2+ influx and cftr activity in g551d and f508del-cftr expressing cells. j cyst fibros. 2013 dec;12(6):584-91
[3] A. Fohner. “PharmGKB summary: ivacaftor pathway, pharmacokinetics/pharmacodynamics.” Pharmacogenetics and genomics 30 1 (2017): 39–42.
[4] Michelle E Condren, & Marquita D Bradshaw. “Ivacaftor: a novel gene-based therapeutic approach for cystic fibrosis.” Journal of Pediatric Pharmacology and Therapeutics 18 1 (2013): 8–13.