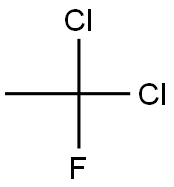
Preparation of Dichlorofluoroethane
Dichlorofluoroethane (HCFC-141b, 1,1-dichloro-1-fluoroethane) is an organic solvent and an important amethylating agent.
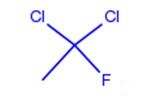
Dichlorofluoroethane can be prepared according to the reported literatures[1-4].
Method 1
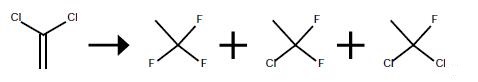
The vapor phase fluorination reaction is conducted in the same 2.54 cm diameter.x.81 cm long Monel.(R). reactor preceded by a vaporizer as in Examples 1 and 2. The reactor is heated with an electric furnace. The reactor is loaded with 100 ml of SbCl5 supported catalyst that is prepared and activated by the same procedures as described in Example 1. The reactor is kept at 100° C. and at atmospheric pressure. The HF flow rate is adjusted to 0.35 g/min and the 1,1-dichloroethylene (VDC) is started at 0.28 g/min. This provides a mole ratio of HF to VDC of about 8:1. The contact time is about 10 seconds. The reaction is run at these conditions until stable reaction conditions are reached. The reaction is monitored by taking reactor effluent samples directly into an in-line GC as described in Examples 1 and 2. The conversion of the VDC is >95percent on a molar basis. The desired HFC-143a product is produced in good yields (>80percent on a molar basis) at these conditions with the majority of the other products being intermediates HCFC-141b and HCFC-142b.
Method 2
The reactions were analyzed using a Hewlett-Packard 5890 gas chromatograph with thermal conductivity detectors, using a 30-foot, 0.5 mm, DB-1 megabore capillary column. Peaks were identified with a Hewlett-Packard 5971A mass selective detector. The experiments were run in a 300 ml stirred Hastaloy C Parr autoclave fitted with a thermocouple and pressure gauge. In the bomb were placed sulfolane and TiCl4 or anhydrous TiF4.
The bomb was sealed and weighed. A weighed quantity of HF was then added through a dip tube at room temperature. A moderate exotherm from the solvation of HF in the sulfolane was observed. When TiCl4 was used, pressure built up in the reactor from liberation of HCl. Next, the bomb was cooled to about 10° C. When HCl was present, the bomb was vented and reweighed.
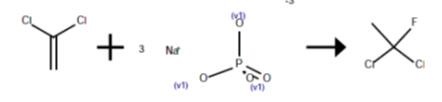
To the cold mixture was added 81 ml (i.e., 97 g or 1 mole) of vinylidene chloride through the gas inlet by means of a syringe. The bomb was then placed in a preheated bath fitted with a temperature controller, and brought to the desire temperature as rapidly as possible, and maintained thereafter at that temperature. Immediately after the desired temperature was reached, and periodically thereafter until the reaction was complete, a vapor sample was withdrawn by attaching a 50 ml polyethylene syringe containing 1-2 g of crystalline trisodium phosphate to the gas outlet.
The syringe was capped and shaken until the HF was neutralized. The vapor sample was then analyzed by gas chromatography. The sampling times were then adjusted to take into account the reaction that took place during the heatup period, and to provide the best fit of the rate constant K for the reaction. The K values thus obtained were subjected to regression analysis. The yield of crude HCFC-141b was determined by distilling the volatiles from the bomb at 60°-70° C. through a dry ice-cooled condenser into a teflon-coated separatory funnel cooled in an ice bath. The upper layer comprised HF, while the lower layer comprised HCFC-141b. The HCFC-141b fraction was separated, washed with water, dried over anhydrous, potassium carbonate, and weighed.
Method 3
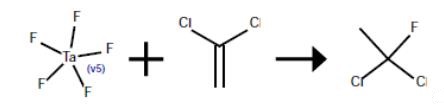
An 80-ml Hastelloy bomb was charged with 2.8 g (0.01 mole) of tantalum pentafluoride. The bomb was evacuated, cooled in dry ice and acetone and charged with 10 g (0.5 mole) of hydrogen fluoride and 38.8 g (0.2 mole) of 1,1-dichloroethene. The mixture was agitated for 3 hr at 25°. The contents of the bomb tube were transferred to an evacuated Hoke cylinder which was cooled in dry ice and acetone. The Hoke cylinder was connected to a vacuum line and the volatile contents were vacuum distilled into a 250-ml polychlorotrifluoroethylene vessel containing 40 g of cracked ice and cooled in liquid nitrogen. This mixture was allowed to melt. The lower organic layer was separated and washed with water to give 16.8 g of 1,1-dichloro-1-fluoroethane; proton NMR δ2.42 ppm (d, J=16.5 Hz). The nonvolatile residue in the Hoke cylinder was 11.9 g of dark oil which appeared to be a complex mixture from its NMR spectrum.
Method 4
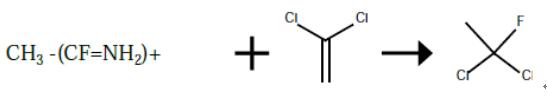
The preparation of 1,1-dichloro-1-fluoroethane from vinylidene chloride
A 300 ml autoclave was charged with 81 g of CH3 --(CF=NH2)+ F- *2HF (1.0 mole). The reactor was evacuated and cooled with a dry ice/acetone bath. 100 g (5.0 moles) of anhydrous HF was charged, followed by 73.5 g (0.5 moles) of vinylidene chloride. The reactor was heated to 50° C. for a period of 2 hours. The reactor was cooled to 20° C. and the product and the product isolated by liquid phase separation as the more dense layer.
References
1. Honeywell International Inc. Method of making difluoromethane, 1,1,1-trifluoroethane and 1,1-difluoroethane. US2005/222472[P], 2005, Page column 3.
2. Laroche Chemicals, Inc. Method for preparing 1,1-dichloro-1-fluoroethane US5336816[P], 1994, A.
3. E. I. Du Pont de Nemours and Company. TaF5 and NbF5 as fluorination catalysts US4258225[P], 1981, A.
4. LaRoche Industries Inc. Production of organic fluorine compounds. US5777185[P], 1998, A.