Trametinib: Potent MEK Inhibitor with Specific Pharmacokinetic and Pharmacodynamic Properties
General Description
Trametinib is a potent reversible allosteric inhibitor of MEK 1 and 2, with specific pharmacokinetic properties. It is rapidly absorbed after oral administration, reaching peak plasma concentration at 1.5 hours, and has an absolute bioavailability of 72%. When taken with a high-fat meal, its exposure decreases, causing a decrease in Cmax and AUC. Trametinib exhibits high plasma protein binding (97%) and is mostly eliminated through feces (80%) and urine (<20%). It has a mean apparent plasma terminal elimination half-life of 3.8 to 4.8 days. Trametinib displays limited drug interactions, but caution is advised when co-administered with drugs metabolized by CYP2C8 and CYP3A4. Pharmacodynamically, trametinib selectively targets MEK1 and MEK2, inhibiting their kinase activity and suppressing downstream ERK signaling. It demonstrates efficacy in targeted cancer therapy, particularly in BRAF and NRAS mutant cell lines.

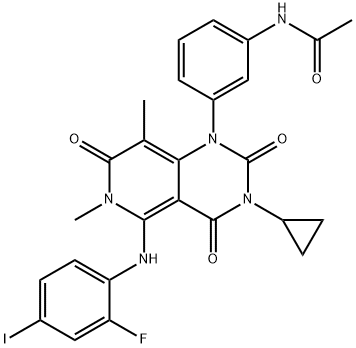
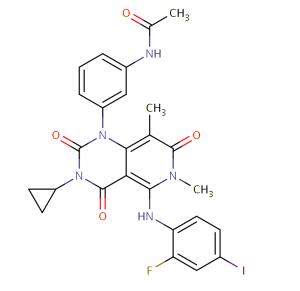
Figure 1. Trametinib
Pharmacokinetics
Trametinib, a potent reversible allosteric inhibitor of MEK 1 and 2, exhibits specific pharmacokinetic properties. Upon oral administration, it is rapidly absorbed with a peak plasma concentration (T max) reached at 1.5 hours and an absolute bioavailability of 72%. In patients receiving a single oral dose of trametinib 2mg per day, the area under the curve (AUC) and mean peak plasma concentrations (Cmax) are measured at 370 ngh/mL and 22.2 ng/ml respectively. When administered with a high-fat meal, trametinib's systemic exposure decreases, leading to a 70% decrease in Cmax and a 24% decrease in AUC. This also prolongs T max to 5.5 hours compared to the fasted state, potentially impacting its efficacy. Therefore, it is recommended to administer trametinib on an empty stomach. Trametinib exhibits high plasma protein binding (97%) and has an apparent volume of distribution (Vd) of 214 L. The drug is mostly eliminated via feces (80%) and urine (less than 20%), with less than 0.1% excreted unchanged. The mean apparent plasma terminal elimination half-life ranges from 3.8 to 4.8 days, following a single oral dose of 2 mg in patients. The mean apparent clearance of trametinib is 4.9 L/h, with trough concentrations ranging from 10.0 to 18.9 ng/mL. 1
Drug interactions
Trametinib, due to its unique metabolic pathways and interactions, exhibits limited drug interactions. It is primarily metabolized via non-cytochrome (CYP450) mediated mechanisms, involving deacetylation through hydrolytic enzymes and glucuronidation. Trametinib is not a substrate for CYP enzymes, P-glycoprotein, BCRP, or certain hepatic uptake transporters at clinically relevant concentrations. However, trametinib may inhibit CYP2C8 and induce CYP3A4 based on in vitro studies. Therefore, caution is advised when co-administering trametinib with drugs metabolized by these enzymes to avoid potential interactions. For instance, dabrafenib, a substrate of both CYP2C8 and CYP3A4, showed a 23% increase in AUC when combined with trametinib, indicating a minor inhibitory effect of trametinib on dabrafenib clearance. Despite this interaction, the recommended daily dosing of the combination remains unchanged due to dabrafenib not being routinely administered at the maximum tolerated dose. 2
Pharmacodynamics
Trametinib is an allosteric kinase inhibitor that selectively targets MEK1 and MEK2, two isoforms of MEK with a crucial role in the MAPK/ERK pathway. By directly binding to unphosphorylated MEK1/2, trametinib inhibits their kinase activity with a high potency characterized by an IC50 of 0.7-0.9 nmol/L. Trametinib's selectivity towards MEK1/2 has been demonstrated by its lack of inhibition towards over 98 other kinases tested. Its pharmacodynamic effects include inhibiting the RAF-mediated phosphorylation and suppressing downstream ERK signaling activity, as evidenced by the decreased levels of phosphorylated ERK, Ki-67 and increased expression of p27. Trametinib has been shown to induce rapid and sustained dephosphorylation of phosphorylated MEK in several cancer cell lines, including BRAFV600E melanoma and HT-29 colon cells, leading to sustained tumor growth inhibition in preclinical models. At a standard dose of 2mg daily, trametinib produces dose-dependent changes in tumor biomarkers, including a median change of 30% inhibition of phosphorylated ERK, 54% inhibition of Ki67 and an 83% increase of p27. Compared with other MEK inhibitors, trametinib exhibits varying binding affinities and differential sensitivity towards mutant cell lines, particularly BRAF and NRAS mutants, highlighting its specificity and efficacy in targeted cancer therapy. 3
Reference
1. Thota R, Johnson DB, Sosman JA. Trametinib in the treatment of melanoma. Expert Opin Biol Ther. 2015;15(5):735-747.
2. Mekinist [package insert]. Research Triangle Park, NC: GlaxoSmithKline; January 2014.
3. Gilmartin AG, Bleam MR, Groy A, et al. GSK1120212 (JTP-74057) is an inhibitor of MEK activity and activation with favorable pharmacokinetic properties for sustained in vivo pathway inhibition [published correction appears in Clin Cancer Res. 2012 Apr 15;18(8):2413]. Clin Cancer Res. 2011;17(5):989-1000.
Related articles And Qustion
See also


Lastest Price from Trametinib manufacturers

US $5.00-0.50/KG2025-06-05
- CAS:
- 871700-17-3
- Min. Order:
- 1KG
- Purity:
- 99% hplc
- Supply Ability:
- 500TONS

US $10.00/KG2024-10-11
- CAS:
- 871700-17-3
- Min. Order:
- 1KG
- Purity:
- 98%
- Supply Ability:
- 10 ton