Synthesis and application of Trimethyloxonium Tetrafluoroborate
Trimethyloxonium Tetrafluoroborate is a white to off-white solid powder under ambient conditions, which is moisture-sensitive and hygroscopic. Primarily employed as a fundamental reagent in organic synthesis, Trimethyloxonium Tetrafluoroborate serves as a strong electrophilic methylating agent, mainly used for the methylation of hydroxyl groups. It finds valuable applications in the total synthesis of marine natural products like spirastrellolide and in the synthesis of pharmaceutical molecules such as lacosamide.
Synthesis

Figure1: Synthesis of Trimethyloxonium Tetrafluoroborate
An oven-dried 500 mL 3-neck round-bottomed flask (marked with a line marked at about 190 mL), equipped with a N2 inlet, 60 mL pressure-equalized dropping funnel, magnetic stirrer bar and rubber septum was placed under a blanket of dry nitrogen gas. DCM (80 mL) was added, followed by boron trifluoride diethyl etherate (33.3 mL, 270 mmol) and the mixture was cooled in a dry ice-acetone bath. Dimethyl ether was condensed into the DCM solution via a needle (through the rubber septum) that remained just below the surface until the total volume of the liquid reached the 190 mL mark. The mixture was stirred vigorously while epichlorohydrin (24.1 mL, 307 mmol) was added drop wise over approximately 15 minutes (the mixture became very thick and required occasion manual swirling to ensure good stirring). The bath was removed and the mixture was stirred vigorously overnight. The resulting solid was collected by filtration through an oven dried, medium frit glass Buchner funnel under a stream of N2 and the flask and filter were rinsed with DCM (2x100 mL). The trimethyloxonium tetrafluoroborate product was isolated as a free-flowing white solid (29.4 g, 98%) after drying under nitrogen and was stored under nitrogen in an oven-dried glass bottle in a freezer. [1]
Reactions with platinum(II) complexes
The reaction of PtCl₂[P(C₂H₅)₃] (1.04 g, 2.0 mmol) with trimethyloxonium tetrafluoroborate (0.56 g, 3.8 mmol) was conducted in 40 mL of dichloromethane under reflux for 9 hours, after which the mixture was cooled and the solvent was evaporated. The resulting residue was then extracted with methanol, and the extract was diluted with an equal volume of diethyl ether, filtered, and treated with hexane until the solution became slightly cloudy. Upon standing, this procedure afforded white needle-like crystals of the known compound Pt₂Cl₃[P(C₂H₅)₃]₂₂ in a yield of 0.60 g (55%), with a measured melting point of 231–234°C, which contrasts with the literature report of decomposition above 250°C without a sharp melting point; nevertheless, the infrared spectrum of the obtained product was identical to that previously reported. [2]
Application
A study has reported the scope and generality of a direct method for the conversion of tertiary amides to methyl esters using Trimethyloxonium Tetrafluoroborate, which involves a two-step, one-pot procedure wherein the tertiary amide is first treated with Trimethyloxonium Tetrafluoroborate to generate an imidate intermediate, followed by hydrolysis typically through the addition of a saturated aqueous sodium bicarbonate solution; although this method is not applicable to aliphatic amides, it affords excellent yields for a variety of amides derived from aromatic carboxylic acids, and steric hindrance at the N-alkyl group is well tolerated, allowing the successful use of N,N-dimethyl, N,N-diethyl, and N,N-diisopropyl amides.
Conversion of tertiary amides to methyl esters
A mild and efficient conversion of various tertiary amides to methyl esters using Trimethyloxonium Tetrafluoroborate has been well documented, wherein the reaction proceeds through the formation of an imidate intermediate upon treatment with Trimethyloxonium Tetrafluoroborate, followed by hydrolysis under basic conditions. While most aryl amides were smoothly transformed into the corresponding methyl esters using this protocol, aliphatic amides and certain ortho,ortho'-disubstituted aryl amides failed to undergo complete hydrolysis, resulting in the regeneration of the starting amides. A notable exception was observed for ortho,ortho'-disubstituted aryl amides where one substituent is a hydroxyl group or a hydroxyl precursor such as a TBS-protected group, which cleaves under the reaction conditions to generate a free phenol. This finding holds significant synthetic relevance, as the resulting aryl triflates—accessible from such phenolic intermediates—can be further functionalized through diverse transformations, thereby offering strategic utility in directed metalation approaches for constructing highly functionalized aromatic systems. Importantly, the steric bulk of the N-alkyl groups exhibited no appreciable effect on the reaction efficiency, enabling the successful conversion of N,N-dimethyl, N,N-diethyl, and N,N-diisopropyl aryl amides into the corresponding methyl esters under these mild conditions. [3]
Reference
[1] O'Connor, Stephen J.; et al, Preparation of imidazodiazepinecarboxamides and related compounds as cannabinoid CB1 and CB2 modulators. United States Patent, Patent Number:WO2010068520.
[2] P.M. Treichel, Reactions of trimethyloxonium tetrafluoroborate and dihalo-bis-ligandplatinum(II) complexes, Inorganica Chimica Acta
1972, 6, 674-676.
[3] Gary E. Keck, ADirect and Mild Conversion of Tertiary Aryl Amides to Methyl Esters Using Trimethyloxonium Tetraˉuoroborate: A Very Useful Complement to Directed Metalation Reactions, Tetrahedron, 2000 56, 9875.
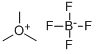
You may like
See also


Lastest Price from Trimethyloxonium Tetrafluoroborate manufacturers

US $0.00-0.00/kg2025-04-24
- CAS:
- 420-37-1
- Min. Order:
- 1kg
- Purity:
- 99
- Supply Ability:
- 20tons

US $0.00/kg2025-04-14
- CAS:
- 420-37-1
- Min. Order:
- 1kg
- Purity:
- 99%
- Supply Ability:
- 10000KGS