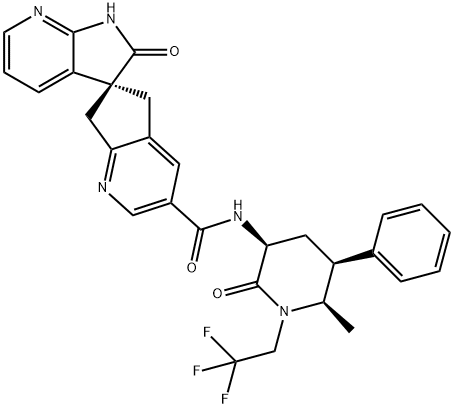
The synthesis method of Ubrogepant
Introduction
Ubrogepant is a first-in-class oral calcitonin gene-related peptide (CGRP) receptor antagonist that Allergen developed under license from Merck & Co. It is an orally administered small-molecule drug that the USFDA approved for the treatment of acute migraine in adults with or without aura. In addition, Ubrogepant earned USFDA approval in February 2020.
Synthesis of Ubrogepant Aminopiperidinone Fragment 140

The route depicted here has been demonstrated on the process scale and features fewer synthetic steps than the original discovery route[1]. The convergent synthetic strategy employed a late-stage amide bond formation to unite two key fragments, 140 and 153. The preparation of aminopiperidinone fragment 140 commenced with L-serine isopropyl ester 132, which was converted to iodide 133 via a Finkelstein reaction. This compound was then subjected to an alkylation reaction with 4-bromophenylacetone (134) and subsequent hydrogenative debromination to furnish keto ester 135 as an undisclosed mixture of isomers. Next, the critical step utilized a transaminase-induced dynamic kinetic resolution to produce piperidinone 136 in high yield with high diastereoselectivity at C-4 and C-5 (dr > 60:1). The diastereomeric ratio at C-2 was approximately 1.2:1. N-trifluoroethylation was effected under strongly primary conditions to obtain the desired monoalkylated intermediate 138 and an undisclosed proportion of bisalkylated product 139. After Boc deprotection, the crude mixture of 138 and 139 was subsequently converted to 4-nitrobenzoic acid salt 140. Crystalline 140 was isolated as a single diastereomer in high yield after 5-nitro-2-hydroxybenzaldehyde-mediated epimerization at C-2.
Synthesis of Ubrogepant Pyrrolidinone 143

Pyrrolidinone 143 was prepared from N-(tert-butyl)-3- methylpyridin-2-amine (141) in two steps (shown above). First, aminopyridine 141 was converted to methyl carbamate 142. Subsequent intramolecular cyclization furnished pyrrolidinone 143.
Synthesis of Ubrogepant Acid Fragment 153

The preparation of acid fragment 153 is described below. First, 2,3-dibromo-5-chloropyridine (144) was treated with i-PrMgCl·LiCl to promote halogen−metal exchange. The aryl magnesium species was then quenched with DMF and reduced to alcohol 145. The hydroxyl moiety in 145 was protected as the TIPS ether 146, and the methyl ester was reduced and converted to benzyl chloride 147. Pyrrolidinone 143 was then alkylated with freshly prepared chloride 147. Subsequent subjection to TBAF generated free alcohol 148. This hydroxyl compound was converted to chloride 149 by treatment with thionyl chloride, setting the stage for the critical intramolecular cyclization reaction. The enantioselective cyclization was accomplished using chiral phase-transfer catalyst 150 to deliver the critical spiroazaindole 151 in high yield and enantiomeric ratio. Chloropyridine 151 was converted to carboxylic acid 152 via palladium-catalyzed carbonylation, and subsequent removal of the tert-butyl group under strongly acidic conditions revealed the key coupling partner 153.
Final Assembly of Ubrogepant

To unite subunits 140 and 153, 4-nitrobenzoic acid salt 140 was treated with aqueous tripotassium phosphate to generate the free-base amine. EDC-mediated amide coupling with carboxylic acid 153 was then delivered as a ubrogepant in a 97% yield over two steps.
References
[1] Andrew C. Flick. “Synthetic Approaches to the New Drugs Approved during 2019.” Journal of Medicinal Chemistry 64 7 (2021): 3604–3657.