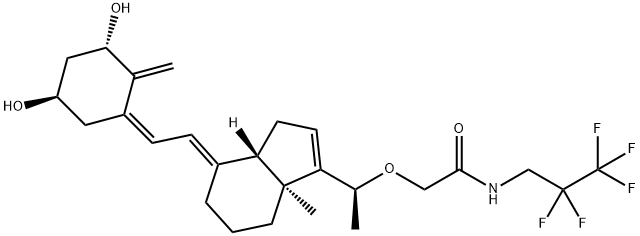
The synthesis method of Tezacaftor
Description
Tezacaftor is a broad-acting CFTR corrector similar to lumacaftor that facilitates the cellular processing and trafficking of normal CFTR and multiple mutant CFTR forms, including F508del, thereby increasing the amount of CFTR protein at the cell surface, resulting in increased chloride transport. Adding a CFTR corrector to potentiator therapy provides clinical benefit to those carrying processing and trafficking mutations such as F508del, the most common CF-causing CFTR mutation[1]. In addition, CFTR corrector and potentiator combination therapy may provide benefits to patients who are compound heterozygous for F508del and an ivacaftor-responsive mutation.
Synthesis method
Synthesis of Tezacaftor Cyclopropylacetic Acid 368

Multiple patent applications have been filed by scientists at Vertex describing the preparation of tezacaftor, all of which outline similar reaction sequences[2]. The route depicted here has been chosen based on a higher reported scale, better efficiency, and fewer step counts. The convergent synthetic strategy utilizes a late-stage amide bond formation to unite two key fragments. Preparation of the acid fragment 368 commenced with the palladium-catalyzed decarboxylative arylation involving ethyl cyanoacetate 365 with aryl bromide 364 in the presence of a bulky phosphine ligand. The reaction yielded benzonitrile 366 in 66% yield after acidification. Cyclopropanation of 366 via double alkylation of ethylene fragment 367 in the presence of a phase transfer catalyst followed by basic hydrolysis delivered carboxylic acid 368, purified by recrystallization from hot toluene.
Synthesis of Tezacaftor

Synthesis of the indole subunit began with regioselective bromination of 3-fluoro-4-nitroaniline (369) to arene 370 (Scheme 67). Lewis acid-catalyzed epoxide ring opening of (R)-glycidyl benzyl ether (371) by aniline 370 followed by reduction of the nitro group furnished hydroxy p-phenylenediamine 372, which was isolated as the p-TsOH salt. The salt was treated with aqueous NaHCO3 in DCM to generate free base 372 before the next step. It should be noted that the yields for conversion of 370 to 372 were not reported by the authors. A Sonogashira coupling between aryl bromide 372 and terminal alkyne 373 (whose synthesis is described in Scheme 68) followed by a Larock-type cyclization completed the preparation of aminoindole subunit 374. To unite key subunits 368 and 374, carboxylic acid 368 was first converted to an acid chloride and then reacted with aminoindole 374 to deliver bis-benzyl-protected tezacaftor. Finally, global deprotection via palladium-catalyzed hydrogenation furnished tezacaftor (XXXVII) in 68−84% yield (based on 374).
Synthesis of Tezacaftor Alkyne 373

The preparation of alkyne 373 is described in Scheme 68. First, propargyl alcohol 375 was converted to propargyl chloride 376 by treatment with aqueous HCl. A Grignard reagent was generated from chloride 376 and subsequently alkylated with benzyl chloromethyl ether 377. Removal of the trimethylsilyl moiety generated the terminal alkyne 373.
References
[1] Scott H Donaldson. “Tezacaftor/Ivacaftor in Subjects with Cystic Fibrosis and F508del/F508del-CFTR or F508del/G551D-CFTR.” American journal of respiratory and critical care medicine 197 2 (2018): 214–224.
[2] Andrew C. Flick. “Synthetic Approaches to New Drugs Approved during 2018.” Journal of Medicinal Chemistry 63 19 (2020): 10652–10704.