Synthesis of Enarodustat(JTZ-951)
Synthesis of Enarodustat
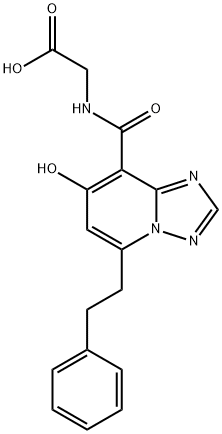
Enarodustat(JTZ-951) is synthesised using Dichloropyridine as a raw material through the reactions of lipo-conversion, substitution, rearrangement, coupling and hydrolysis. The specific synthesis steps are as follows:

Dichloropyridine 62 was converted to ester 64 via olithiation with a subsequent carboxylate trap, followed by Lewis acid-catalyzed esterification employing imidate 63 in 96% yield over two steps. Sequential SNAr reactions involving benzyl alcohol and then hydrazine were followed by treatment with trimethyl orthoformate under acidic conditions to furnish triazolopyridine 65 in 63% yield over three steps. Next, the addition of 65 to morpholine triggered the key Dimroth rearrangement, which was followed by regioselective iodination to afford the rearranged iodinated triazole 66 in 68% yield.
Sonogashira coupling with phentylacetylene followed by removal of the tert-butyl group provided carboxylic acid 67 in 80% yield over two steps. Amide formation with glycine ethyl ester (68) followed by reduction of the alkyne and concomitant removal of the benzyl protecting group liberated the phenol. Ester hydrolysis afforded Enarodustat as the free carboxylic acid in 67% yield over three steps.
Introduction of Enarodustat
Enarodustat was approved by the Japan Pharmaceuticals and Medical Devices Agency (PMDA) in September 2020 for the treatment of anemia associated with chronic kidney disease (CKD). Similar to daprodustat, enarodustat is an orally active inhibitor of HIF-PH and represents an alternative to injectable erythropoietinstimulating agents. Erythropoietin production is regulated by HIF; inhibitorssuch as enarodustat restrict the breakdown of the HIF-α subunit, thereby leading to an increase in endogenous erythropoietin production.