Synthesis of Delgocitinib
Synthesis of Delgocitinib
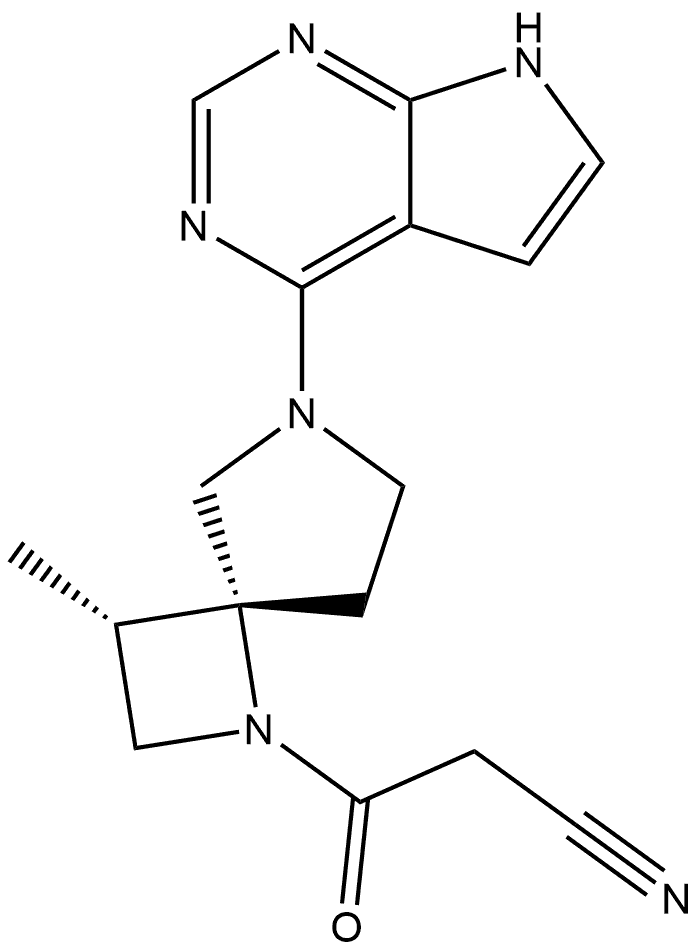
Delgocitinib is synthesised using bromolactone as raw material by chemical reaction. The specific synthesis steps are as follows:

A stereocontrolled kilogram scale synthesis of delgocitinib has been disclosed, beginning with an SN2 reaction involving bromolactone 128 and benzyl amine to provide α-amino lactone 129, which was isolated as the HCl salt after precipitation from hydrochloric acid in ethyl acetate. Amine 129 was then acylated with enantiomerically pure acid chloride 131 (prepared by thionyl chloride treatment of commercial acid 130) to furnish lactone 132. In the crucial spirocyclic ring ringforming sequence of the synthesis, lactone 132 was treated with LHMDS to form an enolate that underwent SN2 displacement of the chloride, forming the spirolactone 133 and establishing both stereocenters with 98:2 dr and 96% ee.
The lactone ring of 133 was then opened by an attack of potassium phthalimide on the γ- carbon, and the resulting carboxylic acid was converted to the ethyl ester by treatment with ethyl iodide. Finally, treatment with diethylenetriamine released phthalimide, providing a free amine for subsequent cyclization to spirolactam 134 via the corresponding ethyl ester intermediate. This sequence took place in 80% yield over four steps and provided the spirolactam in >99% de after recrystallization.
The carbonyl groups within spirolactam 134 were then reduced with lithium aluminum hydride and aluminum chloride in THF, and the resulting diamine 135 was crystallized as a succinic acid salt in 86% yield. The SNAr reaction of 135 with chloropyrrolopyrimidine 136 followed by hydrogenative removal of the benzyl protecting group provided amine 137 in 92% yield over 2 steps. Finally, amine 137 was acylated with cyanoacetyl pyrazole 138 and recrystallized from n-butanol with 3 wt % BHT to provide delgocitinib in 86% yield, >99% ee, and >99% de.
You may like
See also


US $0.00-0.00/mg2025-06-30
- CAS:
- 1263774-59-9
- Min. Order:
- 10mg
- Purity:
- 99%+ HPLC
- Supply Ability:
- 1000