Recalcitrance of 2-Chlorotoluene
ABSTRACT
To elucidate possible reasons for the recalcitrance of 2-chlorotoluene, the metabolism of chloromethylcatechols, formed after dioxygenation and dehydrogenation by Ralstonia sp. strain PS12 tetrachlorobenzene dioxygenase and chlorobenzene dihydrodiol dehydrogenase, was monitored using chlorocatechol dioxygenases and chloromuconate cycloisomerases partly purified from Ralstonia sp. strain PS12 and Wautersia eutropha JMP134. Two chloromethylcatechols, 3-chloro-4-methylcatechol and 4-chloro-3-methylcatechol, were formed from 2-chlorotoluene. 3-Chloro-4-methylcatechol was transformed into 5-chloro-4-methylmuconolactone and 2-chloro-3-methylmuconolactone. For mechanistic reasons neither of these cycloisomerization products can be dehalogenated by chloromuconate cycloisomerases, with the result that 3-chloro-4-methylcatechol cannot be mineralized by reaction sequences related to catechol ortho-cleavage pathways known thus far. 4-Chloro-3-methylcatechol is only poorly dehalogenated during enzymatic processing due to the kinetic properties of the chloromuconate cycloisomerases. Thus, degradation of 2-chlorotoluene via a dioxygenolytic pathway is evidently problematic. In contrast, 5-chloro-3-methylcatechol, the major dioxygenation product formed from 3-chlorotoluene, is subject to quantitative dehalogenation after successive transformation by chlorocatechol 1,2-dioxygenase and chloromuconate cycloisomerase, resulting in the formation of 2-methyldienelactone. 3-Chloro-5-methylcatechol is transformed to 2-chloro-4-methylmuconolactone.
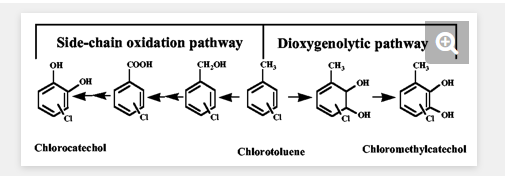
The degradation of chloroaromatics is usually initiated by peripheral enzyme reactions, which activate the aromatic ring and a special chlorocatechol pathway (40). Two distinct peripheral pathways have been described for the metabolism of chlorotoluenes (Fig. 1). Degradation can be initiated by dioxygenation, such that the methyl substituent stays intact and the degradation occurs via the respective chloromethylsubstituted catechols (13). Alternatively, degradation can be initiated via oxidation of the methyl substituent which is subsequently eliminated during further processing reactions, resulting in chlorocatechols as intermediates (2). Pathways via monooxygenation of the side chain of chlorotoluenes have been reported to be functional for 3-chlorotoluene and 4-chlorotoluene as well as 3,5-dichlorotoluene in strains harboring the TOL plasmid-encoded upper pathway for the transformation of the substrates into the respective benzoates and catechols and harboring a chlorocatechol pathway for degradation of chlorocatechols (2). However, due to the restricted substrate specificity of xylene monooxygenase (2) and toluate dioxygenase (11), 2-chlorotoluene (and probably other 2-chlorosubstituted toluenes also) cannot be degraded by such strains, and engineering using a monooxygenolytic pathway has failed thus far (14).
The alternative dioxygenolytic pathway has been shown to be productive for the mineralization of 4-chlorotoluene (2, 13) as well as of 2,4-, 2,5-, and 3,4-dichlorotoluene (34). The failure of the 2,4-, 2,5-, and 3,4-dichlorotoluene-degrading strain Ralstonia sp. strain PS12 to grow with 2-chlorotoluene or 3-chlorotoluene or with 2,3-dichlorotoluene, 2,6-dichlorotoluene, and 3,5-dichlorotoluene was due to the fact that tetrachlorobenzene dioxygenase catalyzed predominantly a monooxygenation of the methyl side chain that results in the misrouting of these substrates into a dead-end pathway (33). Similarly, toluene dioxygenase of P. putida F1 was observed to catalyze predominantly monooxygenation of the side chain of 2- and 3-chlorotoluene (22). The optimization of chlorotoluene degradation by rational engineering of the regioselectivity of toluene dioxygenases to avoid monooxygenation was therefore suggested (35).
MATERIALS AND METHODS
Bacterial strains and growth conditions.Wautersia eutropha JMP 222 (pBBR1 M-I) (32) and Ralstonia sp. strain PS12 (DSMZ 8910) were grown in mineral salts medium (3) containing 50 mM phosphate buffer (pH 7.4) supplemented with 5 mM 3-chlorobenzoate or 1,2,4,5-tetrachlorobenzene as a sole carbon source as fine mortar-ground crystals corresponding to a concentration of 5 mM. Baffled Erlenmeyer flasks (1 to 3 liter) were filled with 100 to 300 ml of medium, sealed with Teflon-coated screw caps, and incubated at 30°C on a rotary shaker (150 rpm).
Escherichia coli DH5α (pSTE44) containing the tetrachlorobenzene dioxygenase tecA1A2A3A4 and chlorobenzene dihydrodiol dehydrogenase tecB open reading frames (33) originating from Ralstonia sp. strain PS12 was grown at 37°C in Luria broth medium (42) containing 1 mM IPTG (isopropyl-a̅-d-thiogalactopyranoside) and 0.1 mg of ampicillin/ml.
For transformation by whole cells, PS12 bacteria were harvested in late exponential growth by centrifugation and resuspended to an A546 of 5 in 10 ml of phosphate buffer (50 mM, pH 7.4). 2-Chlorotoluene was added from a 100 mM stock solution in dimethyl sulfoxide to give a final concentration of 0.3 mM. Flasks were incubated at 30°C on a rotary shaker. For quantification of intermediates, cell-free supernatant fluids were directly analyzed by HPLC analysis and substrate depletion and product accumulation followed for a time period of 2h.
HPLC analyses.Metabolites were analyzed by injection of 10-μl samples. Product formation was analyzed with a Shimadzu HPLC system (LC-10AD liquid chromatograph, DGU-3A degasser, SPD-M10A diode array detector, and FCV-10AL solvent mixer) equipped with a SC125/Lichrospher column (Bischoff, Leonberg, Germany) (5 μm). For analyzing substrate transformation, the aqueous solvent system (flow rate, 1 ml/min) contained 0.01% (vol/vol) H3PO4 (87%) and 50% (vol/vol) methanol as previously described (35). For analysis of metabolites, the methanol concentration was reduced to 25%. Where possible, the metabolites were identified by comparison of retention volume and UV absorption spectra with those of authentic standards.
Mixtures of 3-chloro-4-methyl- and 4-chloro-3-methylcatechol and of 3-chloro-5-methyl- and 5-chloro-3-methylcatechol were prepared from 2-chlorotoluene and 3-chlorotoluene, respectively, by the use of E. coli (pSTE44) as previously described (35).
3-Chloro-, 2-methyl-, 3-methyl-, 2,4-dichloro-, and dichloromethylmuconates as substrates for chloromuconate cycloisomerase were prepared in situ from 4-chloro-, 3-methyl-, 4-methyl-, 3,5-dichloro-, and dichloromethylcatechols by the use of partially purified chlorocatechol 1,2-dioxygenase which was free of any (chloro)muconate cycloisomerase activity. 2-Chloro-4-methylmuconolactone and 5-chloro-3-methylmuconolactones as well as 3-methyl-cis- and 3-methyl-trans-dienelactone were available from previous preparations (30, 39), whereas 2-methyl-cis- as well as 5-methyl-cis-dienelactone (synthesized as previously described) (18, 34) were kindly supplied by Walter Reineke (Chemical Microbiology, Bergische University Wuppertal, Wuppertal, Germany) and Stefan Kaschabek (Interdisziplinäres Ökologisches Zentrum, TU Bergakademie Freiberg, Freiburg, Germany).
2-Methyl-trans-dienelactone was produced by the irradiation of an aqueous solution (2 ml [0.2 mM in a 10-ml beaker]) by the use of an UV lamp and irradiation with light (λ = 254 nm) at a distance of 6 cm for 2 h as described for the preparation of 2-chloro-trans-dienelactone from 2-chloro-cis-dienelactone (19). Reactions were monitored by HPLC. The E210/E260 ratio and the absorption maximum of the product (λmax = 291 nm), which was identical to that of the substrate, confirmed the identity of the expected isomer. All other chemicals were purchased from Aldrich Chemie (Steinheim, Germany), Fluka AG (Buchs, Switzerland), or Merck AG (Darmstadt, Germany).
RESULTS
Transformation of chloromethylcatechols by chlorocatechol 1,2-dioxygenases and chloromuconate cycloisomerases.To elucidate whether chloromethylcatechols can be transformed by chlorocatechol pathway enzymes and, specifically, whether intermediary substituted muconates can be subjected to dehalogenation, a mixture of 3-chloro-4-methylcatechol (8%), 4-chloro-3-methylcatechol (29%), and 2-chlorobenzylalcohol (63%) was prepared from 2-chlorotoluene and a mixture of 3-chloro-5-methylcatechol (2%), 5-chloro-3-methylcatechol (18%), and 3-chlorobenzylalcohol (80%) was prepared from 3-chlorotoluene by the use of E. coli DH5α (pSTE44) containing the tetrachlorobenzene dioxygenase tecA1A2A3A4 and chlorobenzene dihydrodiol dehydrogenase tecB open reading frames (33) originating from Ralstonia sp. strain PS12 as previously described (35) (Fig. 1).
Upon addition of chlorocatechol 1,2-dioxygenase (50 mU/ml) from either strain JMP222 (pBBR1 M-I) or strain PS12 to solutions containing 20 μM 3-chloro-4-methylcatechol plus 75 μM 4-chloro-3-methylcatechol both chloromethylcatechols were rapidly transformed to concentrations < 1 μM in less than 10 min whereas 2-chlorobenzylalcohol remained unchanged. No significant differences were observed between the two chlorocatechol 1,2-dioxygenases, with the overall transformation rate being similar to the rate of 4-chlorocatechol transformation. As evidenced by HPLC analyses, both catechols were transformed simultaneously and the 3-chloro-4-methylcatechol/4-chloro-3-methylcatechol ratio did not change significantly during transformation, indicative of similar specificity constants (kcat/Km values) for 3-chloro-4-methylcatechol- and 4-chloro-3-methylcatechol.
Transformation of 2-chlorotoluene by Ralstonia sp. strain PS12.From the above-described experiments, it can be seen that the amounts of metabolites formed by successive transformation by TecA tetrachlorobenzene dioxygenase, TecB chlorobenzene dihydrodiol dehydrogenase, chlorocatechol 1,2-dioxygenase, and chloromuconate cycloisomerase can be calculated. According to previous results, 8% ± 2% of applied 2-chlorotoluene is transformed into 3-chloro-4-methyl- and 29% ± 2% is transformed into 4-chloro-3-methylcatechols by TecAB (35). 3-Chloro-4-methylcatechol is converted into 2-chloro-3-methyl- and 5-chloro-4-methylmuconolactone in a 3- to 4:1 ratio, indicating that >1% to 2% and 5% to 8%, respectively, of applied 2-chlorotoluene are converted to these products. 4-Chloro-3-methylcatechol is converted into 3-chloro-2-methylmuconolactone and 5-methyl-cis-dienelactone in a 13- to 20:1 ratio, indicating the 1% to 2% and 25% to 30%, respectively, of applied 2-chlorotoluene is converted to these products.
To establish whether the same products were formed in similar ratios by wild-type Ralstonia sp. strain PS12, 2-chlorotoluene (300 μM) was transformed with 1,2,4,5-tetrachlorobenzene-grown cells of this organism. As previously described (22), 2-chlorobenzylalcohol accumulated as an intermediate and was further transformed slowly into 2-chlorobenzoate as a dead-end product. 2-Chloro-3-methyl-, 5-chloro-4-methyl-, and 3-chloro-2-methylmuconolactone accumulated in amounts corresponding to 5 to 8%, <2%, and 20 to 24%, respectively, amounts similar to those expected from experiments with overexpressed and partially purified enzymes. 5-Methyl-cis-dienelactone accumulated in concentrations ≤ 2 μM, indicating that this compound is further transformed by dienelactone hydrolase and possibly mineralized. However, according to the above-presented data, only about 1 to 2% of the applied 2-chlorotoluene is transformed into 5-methyl-cis-dienelactone whereas the rest is transformed into dead-end products.
DISCUSSION
We have previously reported that 2-chlorotoluene is predominantly monooxygenated by tetrachlorobenzene dioxygenase to form 2-chlorobenzylalcohol (22), although more than 30% of the substrate is subject to dioxygenation (35). Similarly, a significant amount of 3-chlorotoluene (approximately 20%) is subject to dioxygenation, resulting mainly in 5-chloro-3-methylcatechol (35). Also toluene dioxygenase of Pseudomonas putida F1 was shown to catalyze both mono- and dioxygenation of 2- and 3-chlorotoluene, indicating that multiple orientations of these substrates in the active site of toluene-chlorobenzene dioxygenases are possible. Usually, chlorosubstituted catechols arising as intermediates after dioxygenation of chloroaromatics are easily degraded by enzymes of chlorocatechol pathways (28, 34, 36, 51). However, as shown here, the presence of an additional methyl function can severely disturb degradation and out of four chloromethylcatechols analyzed only one can be easily degraded and dehalogenated by chlorocatechol pathway enzymes (Fig. 3 and 4).
As suggested previously for the degradation of 4-chloro-2-methylphenoxyacetate, the ring cleavage of 5-chloro-3-methylcatechol (4-chloro-2-methylmuconate) is exclusively subject to 1,4-cycloisomerization to yield 2-methyl-cis-dienelactone as the predominant cycloisomerization product (29, 30). This can be subsequently degraded by dienelactone hydrolase (data not shown). Hence, mineralization of 5-chloro-3-methylcatechol, in contrast to that of 3-chloro-5-methylcatechol, should be rather easily achieved. However, neither 3-chloro-4-methylcatechol nor 4-chloro-3-methylcatechol, as metabolites of 2-chlorotoluene, can be mineralized by enzymes of the chlorocatechol pathways described here and chloromuconate cycloisomerization was determined to constitute the pathway bottleneck. This is clearly evident for 3-chloro-4-methylcatechol metabolism. 3-Chloro-4-methylcatechol is transformed (via 2-chloro-3-methylmuconate) into a mixture of 2-chloro-3-methyl- and 5-chloro-4-methylmuconolactone, indicating that dehalogenation does not occur during 1,4- or 3,6-cycloisomerization. This is not unexpected, as neither of the two dehalogenating mechanisms described for chloromuconate dehalogenation (19, 44, 53) can occur (Fig. 2). It can thus be postulated that the incapability to dehalogenate during 2-chloro-3-methylmuconate cycloisomerization is a feature shared by chloromuconate cycloisomerases.
You may like
Related articles And Qustion


See also
Lastest Price from 2-Chlorotoluene manufacturers

US $10.00/KG2025-04-21
- CAS:
- 95-49-8
- Min. Order:
- 1KG
- Purity:
- 99%
- Supply Ability:
- 10 mt

US $0.00-0.00/kg2025-04-21
- CAS:
- 95-49-8
- Min. Order:
- 1000kg
- Purity:
- 99.8%
- Supply Ability:
- 100000ton