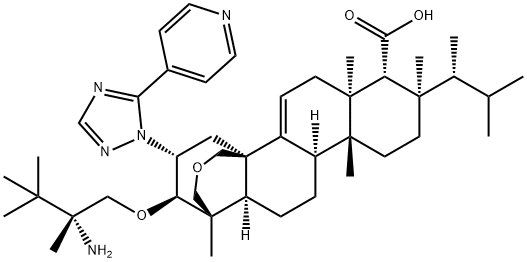
How is Ibrexafungerp synthesised?
Synthesis of Ibrexafungerp
Ibrexafungerp is synthesised in two steps by chemical reaction using enfumafungin as a raw material. The specific synthesis steps are as follows:
Step 1: Preparation of (R)-N-sulfonyl aziridine(Aziridine)
Condensation of 3,3-dimethylbutan-2-one (1.10) with (R)-p-toluenesulfinamide (1.11) gave an 84% yield of compound 1.12, which cyclized upon treatment with trimethylsulfoxonium chloride and n-butyllithium to give chiral toluenesulfinyl aziridine 1.13 in 64% yield. Oxidation of 1.13 with meta-chloroperoxybenzoic acid afforded the tosyl-protected (R)-alpha-disubstituted aziridine 1.5.
-N-sulfonyl aziridine(Aziridine) synthesis")
Step 2: Preparation of Ibrexafungerp
The synthesis of ibrexafungerp started with the natural product enfumafungin (1.1). The lactol of enfumafungin was reduced using triethylsilane and trifluoroacetic acid to give pyran 1.2. Treatment with H2SO4 in methanol resulted in cleavage of the glucose moiety to generate 1.3 in 87% yield over 2 steps. Carboxylic acid 1.3 was converted to the corresponding benzyl ester upon treatment with benzyl bromide to give compound 1.4 in an 89% yield. Reaction of 1.4 with (R)- N-sulfonyl aziridine 1.5 (prepared as shown in Step 1) in the presence of potassium t-pentylate and the cation complexing agent 18-crown-6 provided ether 1.6 in 78% yield. Metal reduction with sodium in liquid ammonia concurrently removed the N-sulfonyl benzyl groups to generate compound 1.7, which was converted to hydrazine intermediate 1.8 with anhydrous hydrazine and BF3·OEt2 in 1,2-dichloroethane (DCE). Cyclocondensation of 1.8 with acyl amidine derivative 1.9 upon heating in acetic acid then provided ibrexafungerp (1) in 66% yield.