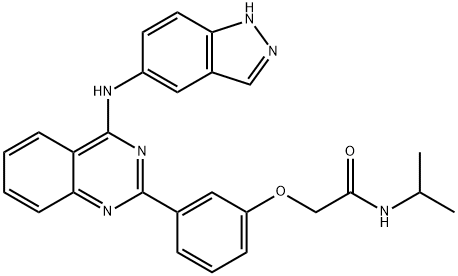
Belumosudil: Synthesis and Description
Synthesis of Belumosudil
Belumosudil is synthesised using bromo acetyl bromide as a raw material by chemical reaction. The specific synthesis steps are as follows:

Reaction of bromo acetyl bromide (17.1) with isopropyl amine (17.2) generated amide 17.3. Subsequently, bromide displacement using methyl 3-hydroxybenzoate (17.4) formed ether 17.5. Ester saponification revealed carboxylic acid 17.6, which was converted to the acyl chloride using oxalyl chloride and then treated with 2-aminobenzamide (17.7) to generate amide 17.8. Base-assisted cyclization resulted in formation of the quinazolinone 17.9, which was then converted to the 4- chloroquinazoline 17.10 using thionyl chloride. The final reaction was a direct SNAr reaction using 5-amino indazole (17.11) that completed the synthesis of belumosidul. The final step to convert belumosudil to the mesylate salt was not described in this patent but likely is achieved by treatment of belumosudil with methanesulfonic acid.
Description of Belumosudil
Belumosudil is an orally active Rho-associated coiled-coil-containing protein kinase-2 (ROCK2) inhibitor recently approved for chronic graft-versus-host disease (cGVHD) after failure of two prior lines of treatment. cGVHD is a chronic inflammatory disease that occurs in 30−70% of patients that undergo allogeneic hematopoietic cell transplantation (allo-HCT). By inhibition of ROCK2, belumosudil lowers cytokine production through downregulation of signal transducer and activator of transcription 3 (STAT3) phosphorylation and promotes regulatory T cell production through upregulation of STAT5 phosphorylation. Developed by Kadmon Pharmaceuticals, belumosudil represents a first-in-class ROCK2 inhibitor and unique mechanism among the recently approved drugs to treat cGVHD.