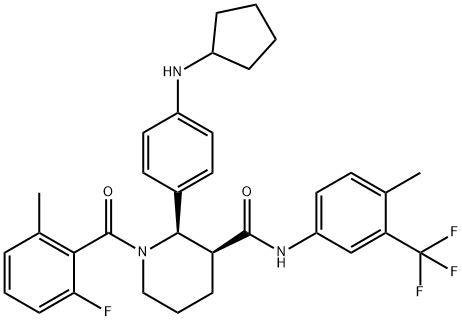
Avacopan: Synthesis and Introduction
Synthesis of Avacopan
Avacopan is synthesised using ethyl 3-(4-nitrophenyl)-3-oxo-propanoate as a raw material by chemical reaction. The specific synthesis steps are as follows:

Exposure of ethyl 3-(4-nitrophenyl)-3-oxo-propanoate 18.1 to acrolein diethyl acetal in the presence of (R)-(−)-2- phenylglycinol 18.2 resulted in the formation of tetrahydropyridine 18.3. Palladium-mediated hydrogenation facilitated stereoselective alkene reduction, presumably deriving from the stereogenicity introduced with 18.2, along with nitro reduction and removal of the bicyclic oxazole fragment to reveal the piperidine. After reductive amination with cyclopentanone, the crude amino ester was obtained in 78% ee. Subsequent salt formation with (−)-O,O′-di-p-toluoyl-L-tartaric acid provided the 1:2 amine:salt adduct 18.4 in 70% yield in >99:1 er from 18.3. Amide 18.6 was subsequently generated following salt breakage and acylation with 2-fluoro-6-methylbenzoyl chloride 18.5. Finally, Lewis acid-mediated amidation with 4-methyl-5- trifluoromethylaniline 18.7 provided avacopan in 80% yield and >99.8% de and ee.
Introduction of Avacopan
Avacopan is a complement 5a receptor (C5aR) antagonist that received its first approval in Japan for the treatment of granulomatosis with polyangiitis (GPA) and microscopic polyangiitis (MPA). GPA and MPA are the two most common forms of anti-neutrophil cytoplasmic autoantibody (ANCA)-associated vasculitis, a systemic autoimmune disease. Avacopan was developed by ChemoCentryx, now acquired by Amgen, and has been approved in the US for adults with severe active ANCA-associated vasculitis (MPA and GPA) as an adjunct therapy.