
一、EGFR基础概述
EGFR又称ErbB1、HER1,隶属于HER(ERBB)受体家族,家族成员共4种:EGFR(HER1)、HER2、HER3、HER4,该家族广泛调控细胞生理活动。 EGFR属于跨膜酪氨酸激酶型糖蛋白,分子量170KDa,广泛表达于上皮细胞、成纤维细胞、胶质细胞、角质细胞表面。EGF、TGFα等配体与细胞膜表面EGFR结合后,受体单体发生同源二聚化,亦可与HER家族其他受体形成异源二聚体,激活胞内激酶区域,启动下游信号级联。当EGFR发生基因突变或蛋白过表达时,会持续驱动肿瘤发生发展。 当前靶向EGFR的小分子TKI、单克隆抗体、双特异性抗体、ADC药物研发管线数量庞大,但治疗后耐药已成为限制临床疗效的核心难题。
二、EGFR完整蛋白结构
EGFR基因定位于人类7号染色体短臂p12-13区域,包含28个外显子,编码1186个氨基酸的跨膜蛋白,整体分为三大结构区域:胞外N端结构域、跨膜疏水α螺旋区、胞内C端结构域。
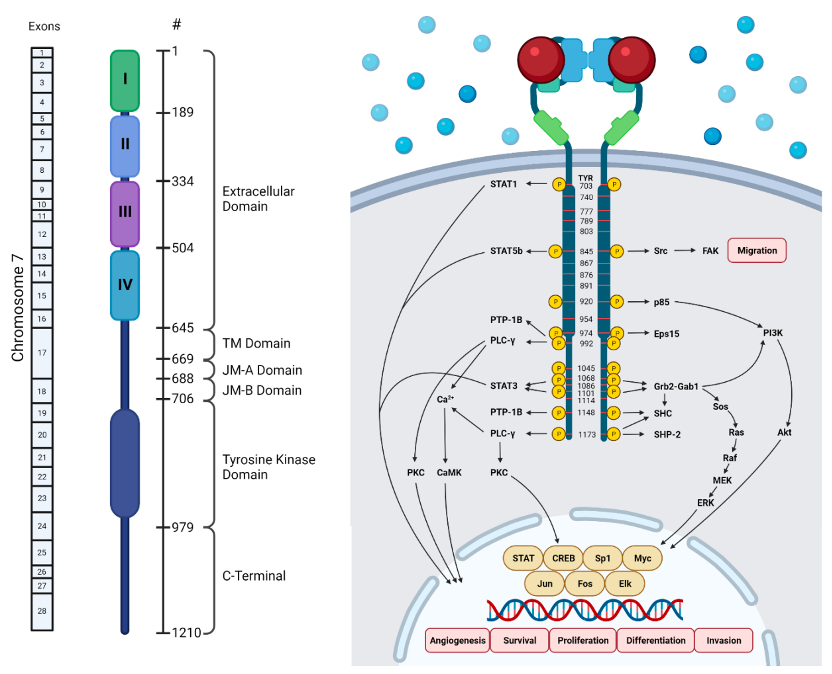
图1. EGFR结构示意图
(1)胞外结构域(622个氨基酸) 分为4个亚结构域:I(1–133位)、II(134–312位)、III(313–445位)、IV(446–621位)。L1(I)、L2(III)为富含β螺旋的亮氨酸结构域,负责结合生长因子;CR1(II)、CR2(IV)为富含二硫键的半胱氨酸区,CR1介导受体同源/异源二聚体形成。L1、CR1、L2共同构成C型结合口袋,容纳配体EGF。
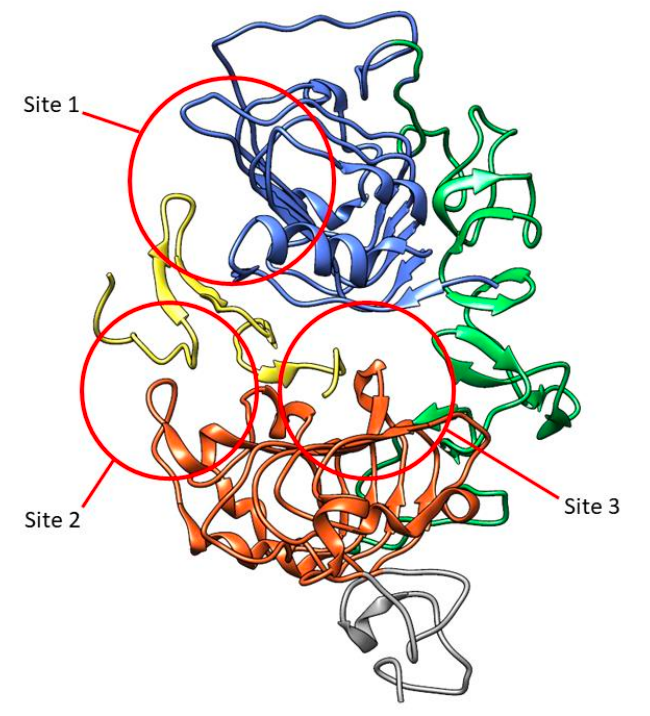
图2. EGFR胞外结构域
EGFR胞外结构域与EGF复合物的三维可视化(黄色)。子域用颜色标记:L1,蓝色;CR,绿色;L2,橙色;部分CR2,灰色的。与EGF相互作用的三个位点用红圈标出。
(2)胞内结构域(542个氨基酸) 包含近膜段(约50个氨基酸)、酪氨酸激酶区(250个氨基酸)、C端尾部(229个氨基酸),C端携带5个自磷酸化位点。 配体结合胞外位点后,EGFR二聚体构象改变,激酶结构域利用ATP发生自磷酸化,进而激活下游多条促癌信号通路。
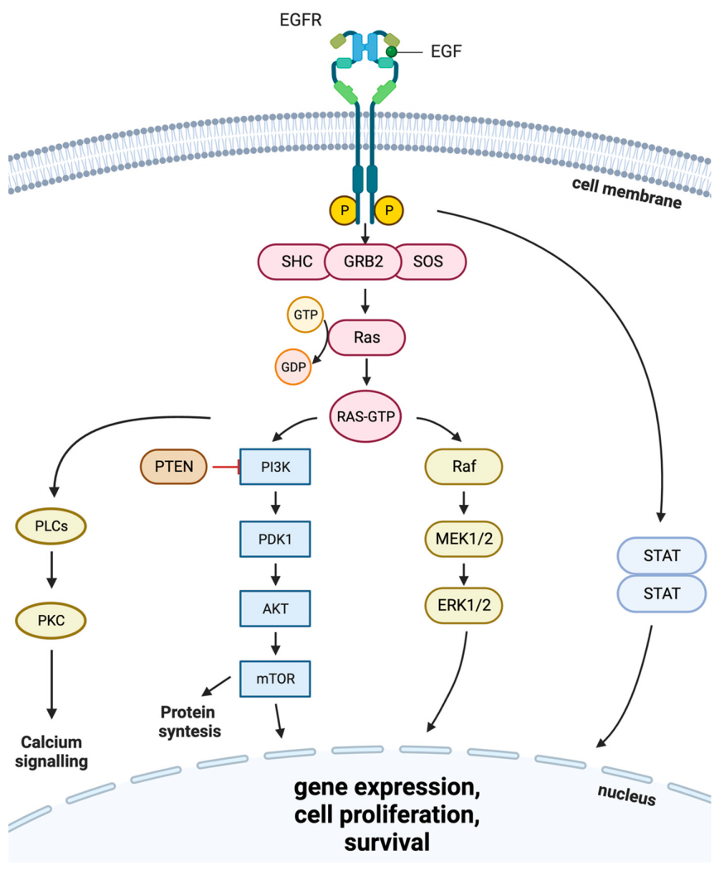
图3. 涉及EGFR的细胞内信号转导
三、EGFR下游核心信号通路
EGFR异常激活后,三条核心通路持续活化,共同推动肿瘤增殖、侵袭、抗凋亡:
(1)Ras/Raf/MEK/ERK1/2通路 EGFR磷酸化后经GRB2、Shc招募SOS蛋白,激活Ras,逐级启动Raf-MEK-ERK级联反应,调控细胞周期与肿瘤增殖。
(2)JAK/STAT通路 促进STAT蛋白入核,激活增殖相关基因转录,加速细胞分裂、抑制肿瘤细胞凋亡。
(3)PI3K/Akt通路 主要介导肿瘤细胞存活,抵抗细胞凋亡。 除此之外,异常活化的EGFR还可联动整合素通路,激活基质金属蛋白酶,削弱细胞粘附能力,促进肿瘤侵袭与远处转移。诱发通路持续激活的诱因包括EGFR基因突变、基因拷贝数扩增、蛋白过表达。
四、EGFR突变与靶向耐药机制
(一)EGFR与非小细胞肺癌(NSCLC)
肺癌为全球致死率最高的恶性肿瘤,我国发病率、死亡率居全部肿瘤首位,其中85%为非小细胞肺癌。EGFR突变是NSCLC最主要的驱动基因突变,亚洲肺腺癌人群突变率超50%,是靶向治疗核心生物标志物。
(二)NSCLC中EGFR突变分型
2004年首次发现NSCLC酪氨酸激酶区体细胞突变,以19外显子缺失、21外显子L858R点突变最为常见,按突变类型分为三类:
I类:外显子19插入缺失突变,缺失E746–S752区间4–6个氨基酸,占全部激酶区突变85%–90%;
II类:18–21外显子单氨基酸替换,以L858R为主;
III类:20外显子重复/插入突变。 另有罕见22外显子E884K突变,会降低各类EGFR抑制剂敏感性。胞内区域突变会造成受体构象不稳定、激酶持续激活,通过抑制凋亡驱动肿瘤进展。
(三)EGFR靶向药获得性耐药机制
(1)T790M守门突变(一代TKI最主要耐药原因)
790位苏氨酸突变为甲硫氨酸(T790M),位于ATP结合口袋入口,更大的氨基酸侧链产生空间位阻,阻碍吉非替尼、厄洛替尼等可逆TKI结合,约50%一代药耐药患者检出该突变。 该突变对不可逆抑制剂(EKB-569、HKI-272)仍敏感,此类药物可与激酶区Cys797形成共价键。临床多见L858R/T790M双突变,双重突变会降低ATP与激酶结合亲和力,耐药程度显著高于单一敏感突变。 2. 非T790M继发突变(代表:D761Y) D761Y常伴随L858R共存,相较单纯L858R对吉非替尼耐药更强,但弱于L858R/T790M;同时对不可逆抑制剂HKI-272敏感性下降,机制为改变EGFR构象、干扰药物结合。 3. 其他旁路耐药 奥希替尼耐药后常见MET/EGFR扩增、KRAS/BRAF突变等旁路激活,占耐药病例约52%。
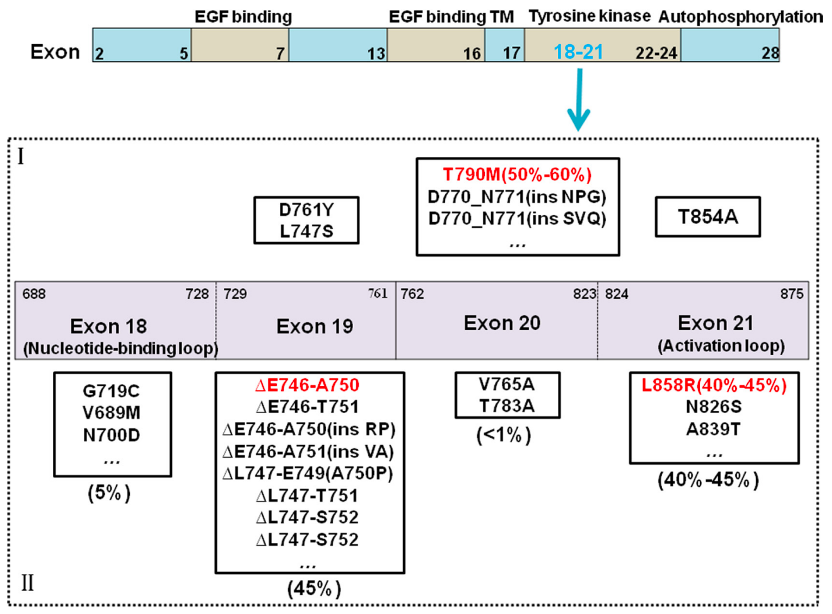
图4.EGFR突变及耐药机制
五、EGFR靶向治疗药物体系
临床靶向EGFR药物分为两大类:胞内小分子酪氨酸激酶抑制剂(TKI)、胞外阻断型单克隆抗体,另有双抗、ADC、CAR-T等新型生物药处于临床开发阶段。
(一)EGFR-TKI小分子抑制剂
TKI可穿透细胞膜,竞争性结合ATP结合位点,阻断突变EGFR持续促癌信号,体外可诱导肿瘤凋亡、抑制血管生成,同时能增敏放化疗、内分泌治疗。依据药物结合特性分为四代:
(1)第一代EGFR-TKI:吉非替尼、厄洛替尼、埃克替尼
特点:可逆结合、无选择性,同时抑制野生型与突变型EGFR。
未筛选人群客观缓解率(ORR)仅10%–20%,中位无进展生存期(PFS)3个月;但在亚洲、非吸烟肺腺癌人群中疗效显著提升,针对19外显子缺失、L858R突变ORR可达75%,中位PFS 7–12个月。 局限:多数患者1年内出现获得性耐药,60%耐药原因为T790M突变。
(2)第二代EGFR-TKI:阿法替尼
特点:不可逆共价结合、广谱抑制全部ErbB家族受体,不区分突变/野生型。
相比一代TKI可延长患者中位总生存期(OS)约8个月,优势来源于广谱阻断HER家族,无法克服T790M耐药。
(3)第三代EGFR-TKI:奥希替尼
特点:不可逆、高选择性,同时覆盖18/19/21外显子敏感突变与T790M耐药突变。
药物转运蛋白亲和力低,血脑屏障穿透性优异,适合合并脑转移患者。2015年FDA获批用于一代/二代TKI进展后T790M阳性NSCLC;基于FLAURA研究获批一线治疗EGFR敏感突变晚期NSCLC。
临床数据:FLAURA研究中奥希替尼一线中位PFS 18.9个月,显著优于一代TKI的10.2个月,降低中枢进展风险,现已成为EGFR阳性NSCLC标准一线方案。
(4)第四代EGFR-TKI(临床研发阶段)
开发目标:克服奥希替尼后继耐药,采用变构抑制机制,不竞争ATP结合位点,通过改变EGFR空间构象阻断信号。代表化合物EAI045、JBJ-04-125-02、BLU-945,对多重耐药突变具备体外活性。
(二)靶向EGFR生物制剂
单克隆抗体结合EGFR胞外结构域,阻断配体结合、抑制受体二聚化,诱导肿瘤细胞凋亡。
(1)已上市单抗适应症:西妥昔单抗、帕尼单抗用于结直肠癌;尼妥珠单抗用于头颈部肿瘤;海外仅耐昔妥珠单抗获批NSCLC。
(2)在研管线(Insight数据库):全球共161个EGFR生物药项目,含33个单抗、30个CAR-T、27个双特异性抗体、21个ADC、4个三特异性抗体。
(三)EGFR/c-MET双靶点药物
MET异常是EGFR-TKI重要旁路耐药驱动因素,在多类肿瘤中高表达:
(1)MET 14外显子跳跃突变:肺腺癌发生率3%–4%;
(2)MET基因扩增:一/二代TKI耐药后5%–22%,三代奥希替尼耐药后15%–30%;
(3)MET蛋白过表达:肿瘤人群占比25%–75%。 全球多数EGFR/c-MET双抗处于早期临床,仅强生Amivantamab获批上市,国内同步申报上市。
六、全球EGFR靶向药物整体研发进展
(1)适应症覆盖:临床阶段药物主要布局非小细胞肺癌、结直肠癌、胃癌、头颈部鳞癌、胰腺癌、食管癌;
(2)上市药物规模:全球共32款EGFR靶向药上市,国内获批22款;其中小分子化药20款,以TKI为主;
(3)肺癌领域布局:全球18款EGFR靶向药获批NSCLC适应症,国内获批14款,是EGFR药物最核心应用赛道;
(4)单抗药物格局:现有上市单抗适应症集中于消化道、头颈部肿瘤,NSCLC可用单抗选择有限。
七、总结与展望
当前EGFR靶向治疗最大瓶颈为治疗后继发耐药,各类新发、罕见突变持续降低TKI敏感性,缩短患者总生存期,同时药物不良反应也需持续优化。 后续研发存在两大核心方向:一是优化胞内酪氨酸激酶抑制剂,开发第四代变构TKI,覆盖多重耐药突变;二是深挖EGFR胞外结构域靶点,拓展单抗、双抗、ADC等生物药应用。此外,TKI与MET抑制剂、化疗、免疫治疗联合用药,多靶点协同克服异质性耐药,将成为未来晚期EGFR突变肿瘤的主流治疗策略。
八、EGFR 肿瘤靶点相关检测技术服务哪里有?
