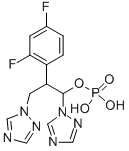
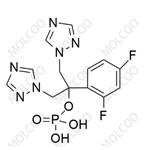
Fosfluconazole is a phosphate prodrug of fluconazole, and it was launched in Japan as an intravenous injection for the treatment of candidiasis and cryptococcosis
infections. Fluconazole, a triazole antifungal agent, is a selective inhibitor
of fungal cytochrome P450 sterol C-14 alpha-demethylation, and it is widely used
for the treatment of patients with serious systemic fungal infections. Fluconazole is
marketed in both oral and intravenous formulations, the latter being a dilute (2 mg/
mL) infusion in saline due to the relatively poor water solubility of the drug. In
patients needing high doses (>400 mg) of fluconazole, a drawback of the IV formulation
is the requirement of a high-volume infusion, which is undesirable in
critically ill patients in whom fluid overload must be avoided. Fosfluconazole is a
prodrug with approximately 40-fold higher water solubility than fluconazole,
thereby achieving a substantial reduction in infusion volume. It is prepared in three
steps starting from fluconazole. In the first step, fluconazole is converted to its
dibenzyl phosphite derivative by reaction with phosphorous trichloride and benzyl
alcohol. Subsequent oxidation of the phosphite to the corresponding phosphate
with hydrogen peroxide and cleavage of the benzyl protecting groups by hydrogenolysis
affords fosfluconazole. In vitro, fosfluconazole is at least 25-fold less potent
than fluconazole against single isolates of Candida species and Cryptococcus
neoformans. In vivo, it is rapidly hydrolyzed to fluconazole by phosphatase enzymes
and exhibits similar efficacy to fluconazole in experimental models of fungal disease.
The hydrolysis potential of fosfluconazole was initially demonstrated in homogenates
of kidney, lung and liver of rat, dog, and human. Subsequently, in clinical trials
with healthy volunteers (n=24), fosfluconazole was shown to hydrolyze rapidly
and almost completely to provide a 97% mean bioavailability of fluconazole. Less
than 1% of the administered dose of fosfluconazole was excreted unchanged in the
urine, with the majority (85.6%) of the dose eliminated as fluconazole. The terminal
half-life was about 2.3 hours, and the volume of distribution was 0.2 L/kg. The time
to reach steady state drug levels with 500 mg daily dose was about 10 days, which
could be shortened to 3 days by administering loading doses of 1000 mgs on days 1
and 2 followed by 500 mg daily. Further studies showed that hepatic or renal
impairment did not significantly alter the pharmacokinetic profile of fosfluconazole.
In phase III studies in patients with deep-seated mycosis due to Candida or
Cryptococcus (n=160), a 2-day loading dose regimen of fosfluconazole provided efficacy range of 73.8% (in Japanese patients) to 91.7% (patients of non-Japanese
origin). The adverse events seen in these trials were similar to those previously
known with fluconazole therapy and included rash (3.1%), abnormal liver function
values (2.5%), asthma (1.9%), and lightheadedness (1.9%).