缬更昔洛韦盐酸盐
发布日期:2019/3/15 11:06:13
背景及概述[1][2]
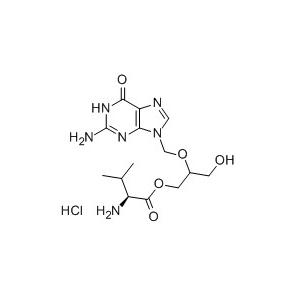
盐酸缬更昔洛韦(valganciclovir hydrochloride),化学名为( S) -2- 氨基-3- 甲基丁酸2-[ ( 2- 氨基-1,6- 二氢-6- 氧代-9H- 嘌呤-9- 基) 甲氧基]-3-羟基丙酯盐酸盐,是瑞士罗氏公司研发的口服抗巨细胞病毒(CMV) 感染药物,2001 年5 月经美国FDA 批准上市。临床用于治疗获得性免疫缺陷综合症(AIDS) 患者因感染CMV 所致急性视网膜炎,2003 年5 月扩大了其适应证,用于预防和治疗器官移植者继发CMV 感染。
该药是更昔洛韦(ganciclovir)的前体药物,是一种活性更昔洛韦缬氨酸酯,口服后可在肠道和肝脏细胞中被磷酸酯酶迅速水解成更昔洛韦,其抗病毒谱和作用机制类同于更昔洛韦,但是它的生物利用度却比更昔洛韦显著提高,其口服吸收的生物利用度为62.4 %,是更昔洛韦的10 倍,而毒性却大大降低。近年来,更昔洛韦保持较高的市场份额,自2002 年起一直名列抗病毒药品市场销售额第二位。若能研究出缬更昔洛韦的生产工艺,则势必会取代更昔洛韦的市场地位,具有广阔的市场前景。
结构
药理作用[3]
缬更昔洛韦是DNA多聚酶抑制剂。其口服后在肠粘膜细胞酯酶和肝酯酶的作用下迅速水解成更昔洛韦,因此其药效学特性即更昔洛韦的药效学特性。更昔洛韦在病毒内和细胞内酶磷酸化作用下生成三磷酸更昔洛韦,后者与三磷酸脱氧鸟苷(dGTP)竞争作为病毒DNA 多聚酶的底物,因此抑制病毒DNA 的合成,从而产生抗CMV 活性。更昔洛韦的体外抗CMV 活性为阿洛韦的26 倍,其IC50为0 .6 ~ 4 .9 μmol·L-1 ,平均2 .7 μmol·L-1 。
临床评价[3]
一项随机、非盲研究比较了缬更昔洛韦和更昔洛韦诱导治疗新感染的AIDS 相关CMV视网膜炎的有效性。160 例病人随机口服缬更昔洛韦900 mg 或静注更昔洛韦5 mg·kg-1 ,bid ,3 w k 后改为qd ,再治疗1 w k 。4 w k 后所有病人口服缬更昔洛韦(900 mg ,qd)进行维持治疗。治疗前各组病人的CD+4 细胞计数和高活性抗逆转录病毒治疗(HAART)情况均相似。
诱导治疗的第1 终点为治疗4 wk 时CMV 视网膜炎病情进展,第2 终点为CMV 视网膜炎进展时间和取得满意疗效。口服缬更昔洛韦组和静注更昔洛韦组,CMV 视网膜炎病情进展的病人数相似,均为10 %。口服缬更昔洛韦组比静注更昔洛韦组的病情进展中位时间长,分别为160 d 和125 d ,而病情进展平均时间相似,分别为226 d 和219 d 。口服缬更昔洛韦和静注更昔洛韦诱导治疗4 w k 时取得满意疗效的比例相似,分别为72 %和77 %。治疗开始和治疗4 wk 时CD+4 细胞计数、CD+4 细胞计数增值和HAART 等亚组数据分析表明,缬更昔洛韦和更昔洛韦的疗效相似。
适应症及常用剂量[4]
包装:0.45g/片。
CMV视网膜炎: 诱导期,900mg,po,q12h,疗程3 周。维持期,900mg,po,qd,直至免疫重建 (连续3~6个 月CD4>150/μL,眼科检查证实病变稳定)。
胃肠道CMV病: 900mg,po,q12h,疗程3~6周 (病情严重或复发时考虑维持治疗)。
接受肾脏、心脏和肾脏—胰腺联合器官移植的CMV感 染高危患者的预防: 900mg,po,qd,自移植10天内开始,直至移植后100天。
禁忌证[4]
对缬更昔洛韦、更昔洛韦或药品中任何其他 成分有过敏反应者禁用。严重粒细胞缺乏 (ANC<500/μL)、 血小板减少 (<25 000/μL)、贫血 (血红蛋白<80g/L) 及肾衰竭者禁用。
副作用[4]
常见可逆性粒细胞减少 (对G-CSF反应好)、 血小板减少、腹泻、恶心。偶见贫血、发热、皮疹、头痛、 头晕。罕见肝毒性、抽搐。
注意事项 [4]
盐酸缬更昔洛韦是更昔洛韦的前体药物,口服后迅速转化 成更昔洛韦,需与食物同服。用药期间需监测血象;外周血中性粒细胞计数<500/μL停药或加用G-CSF; 外周血血小板计数<25 000/μL或血红蛋白<80g/L考虑停药;有中枢 神经系统副反应者避免驾驶汽车和操作机械。
药物相互作用[4]
由于缬更昔洛韦迅速且完全转化为更昔洛韦,因此与更昔洛韦有相互作用的药物与缬更昔洛韦也有相互作用,包括骨髓抑制剂、肾毒性药物、丙磺舒(probenecid)、齐多夫定(zidovudine)和去羟肌苷(didanosine)等。
骨髓抑制剂与更昔洛韦合用能增加后者的血液毒性,如齐多夫定、MMF 或硫唑嘌呤(azathioprine)与缬更昔洛韦合用,使中性粒细胞减少和贫血等不良反应增加;肾毒性药物损害肾功能,使更昔洛韦的体内消除减慢,药物蓄积,毒性增加;丙磺舒和其他肾排泄药物能降低更昔洛韦的清除率,也导致其毒性增加;更昔洛韦可显著增加去羟肌苷的生物利用度(22 %~ 110 %),应密切观察后者引起的毒性反应。
制备 [1]
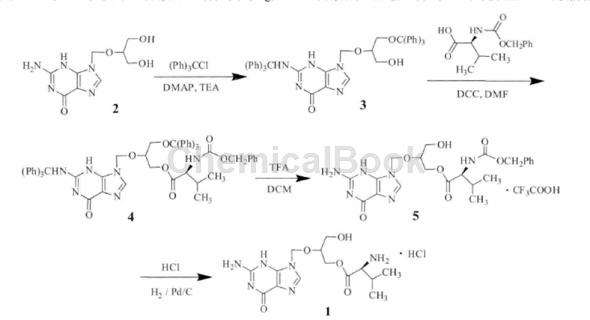
2 与三苯基氯甲烷在缚酸剂存在下对氨基和羟基进行保护,可得到3 种取代物,利用其性质差异,将反应液冷却后抽滤可除去难溶的三取代物;滤液倒入水中,去除易溶于水的一取代物,过滤并多次洗涤得到二取代物3,纯度98.1%。3 再与N- 苄氧羰基-L- 缬氨酸缩合制得4,替换文献中不易得到的(S)-3- 苄氧羰基-4- 异丙基-2,5- 噁唑烷二酮。
4在三氟乙酸的二氯甲烷溶液中脱除三苯甲基得5,用二氯甲烷替换了价格较贵的2,2,2- 三氟乙醇。5溶于甲醇,加盐酸和10%钯炭,通入氢气常压氢化,最后经异丙醇和水重结晶得到盐酸缬更昔洛韦。改进后盐酸缬更昔洛韦的总收率为34%,反应条件温和,操作简便,成本降低,适合工业化生产。
主要参考资料
[1] 盐酸缬更昔洛韦的合成
[2] 新型抗病毒药物缬更昔洛韦的合成综述
[3] 口服抗病毒新药—缬更昔洛韦
[4] 协和抗感染手册