甲磺酸达比加群酯的制备方法和有关物质检测
发布日期:2020/7/6 11:16:11
背景及概述[1]
甲磺酸达比加群酯是应用于临床的直接凝血酶抑制剂,是一种新型口服抗凝药。其作用机理是通过可逆性强效竞争结合凝血酶的纤维蛋白特异结合位点,使纤维蛋白生成受阻,从而抑制血栓的形成。甲磺酸达比加群酯是达比加群的前体药物,在体内代谢后转化为具有活性的达比加群。与华法林相比,甲磺酸达比加群酯在治疗过程中无需频繁监测凝血功能和调整给药剂量,药物之间相互作用少,不受进食影响,从而提高了患者的用药依从性。
有关物质检测[1]
强文舟等人参考国家食品药品监督管理局达比加群酯胶囊进口药品注册标准关于有关物质的规定,并结合收敛型原料药合成工艺,共获得9个已知杂质(A~I,结构见图1),其中杂质A、B、C和D为合成工艺杂质,其余为降解杂质。建立了甲磺酸达比加群酯原料药中有关物质HPLC检测法,本方法适用性好试验效率高,通过验证表明此方法专属性强且灵敏度高,能对甲磺酸达比加群酯中有关物质进行有效控制。
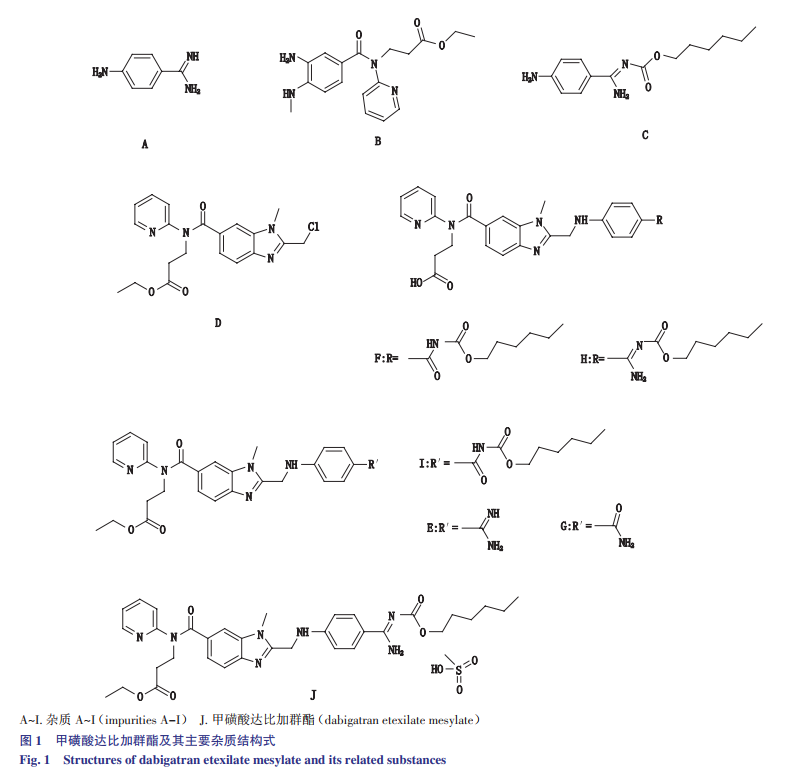
1仪器、样品与试剂
1.1仪器
岛津公司LC-2010AHT高效液相色谱仪,SPDM20A二极管阵列检测器,LabSolution色谱工作站;百万分之一天平(赛多利斯公司)。
1.2样品与试剂
甲磺酸达比加群酯原料药(批号140301、140302、140303)、甲磺酸达比加群酯对照品(纯度≥99.5%,批号140204),西安新通药物研究有限公司;对照品杂质A(纯度≥98%,批号14011202)、杂质B(纯度≥98%,批号14011902),上海百谊生物科技有限公司;对照品杂质C、杂质D为合成工艺中间体,纯度均≥98%,西安新通药物研究有限公司;对照品杂质E(纯度≥99.5%,批号D01833)、杂质F(纯度≥99.5%,批号D01835)、杂质G(纯度≥99.5%,批号01836)、杂质H(纯度≥99.5%,批号D01837)、杂质I(纯度≥99.5%,批号D01834),深圳菲斯生物科技有限公司;乙腈为色谱纯,其他试剂均为分析纯。
2方法与结果
2.1色谱条件
采用菲罗门PhenomenexC18(250mm×4.6mm,5µm)色谱柱,以pH4.5的醋酸铵缓冲液(A)-乙腈(B)为流动相,梯度洗脱(程序见表1),流速1.0mL·min-1,柱温为40℃,检测波长为242nm(检测杂质B、D、G)、310nm(检测杂质A、E、F、I)、340nm(检测杂质C、H),进样量10µL。
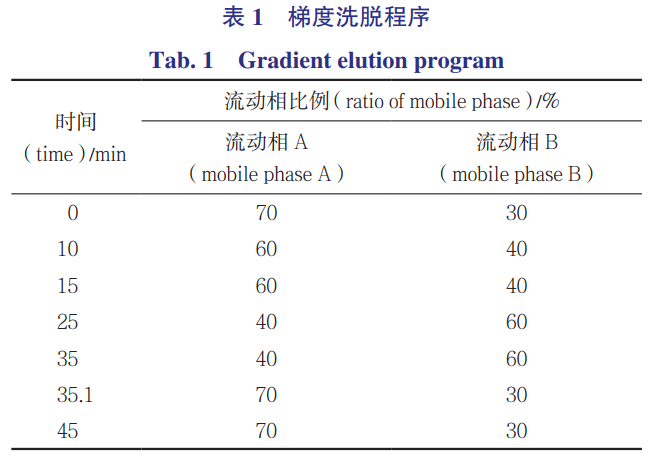
2.2系统适用性试验
取杂质A~I的对照品各5mg,精密称定,分别置25mL量瓶中,加30%乙腈溶解并稀释至刻度,摇匀,即得各杂质的储备液。取甲磺酸达比加群酯原料药10mg,精密称定,置10mL量瓶中,加30%乙腈约5mL溶解,再分别精密加入各杂质的储备液100µL,加30%乙腈稀释至刻度,摇匀,即得杂质定位混合溶液;进样10µL,记录色谱图,见图2。杂质A~I各峰之间的分离度均大于1.5,达比加群酯与相邻杂质峰之间的分离度大于1.5。
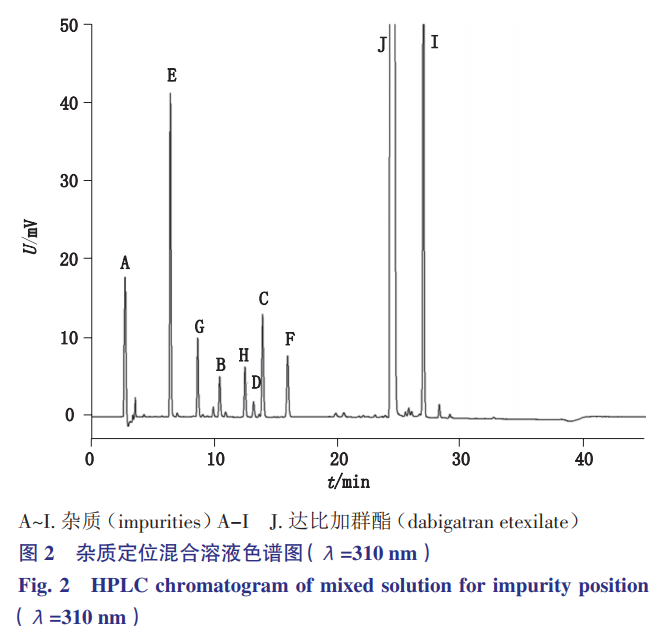
2.3线性关系考察
分别取各杂质储备液及甲磺酸达比加群酯对照品适量,加30%乙腈配制成1mL中含杂质A、B、C、D各1μg的混合溶液1,分别进样6、8、10、12、15μL;1mL中含杂质E、F、G、H各2μg的混合溶液2,分别进样3、10、15、20、30、40μL;1mL中含杂质I、达比加群酯各4μg的混合溶液3,分别进样1、3、10、15、30、50、80μL。以进样量(ng)为横坐标,峰面积为纵坐标,进行线性回归,得回归方程。同时计算各杂质相对于达比加群酯的校正因子。结果见表2。
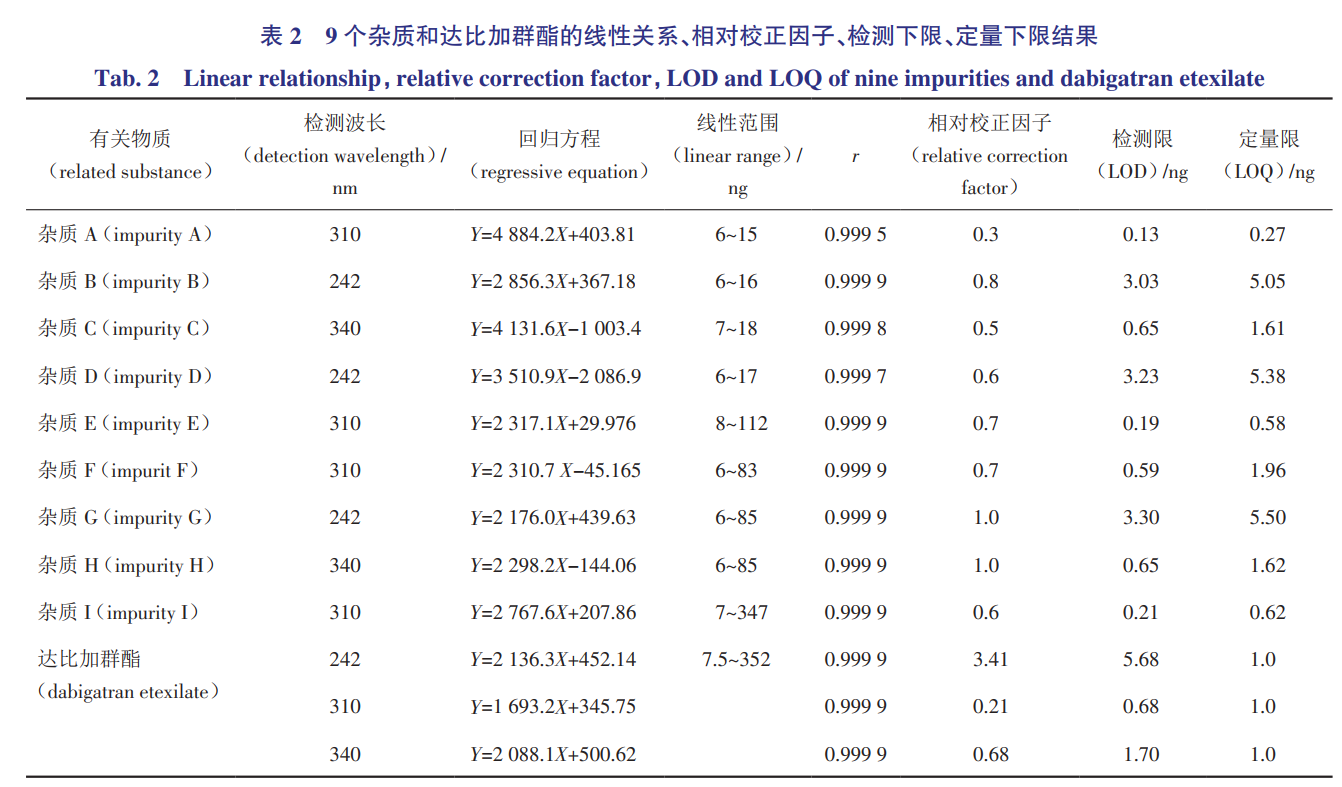
2.4检测下限和定量下限
称取杂质A~I及甲磺酸达比加群酯的对照品适量,分别加30%乙腈配制成1mg·mL-1的储备液,并稀释制成系列浓度溶液,取10μL注入液相色谱仪,记录色谱图。以信噪比S/N≥3时的测定结果作为检测限,信噪比S/N≥10时的测定结果作为定量限见表2。
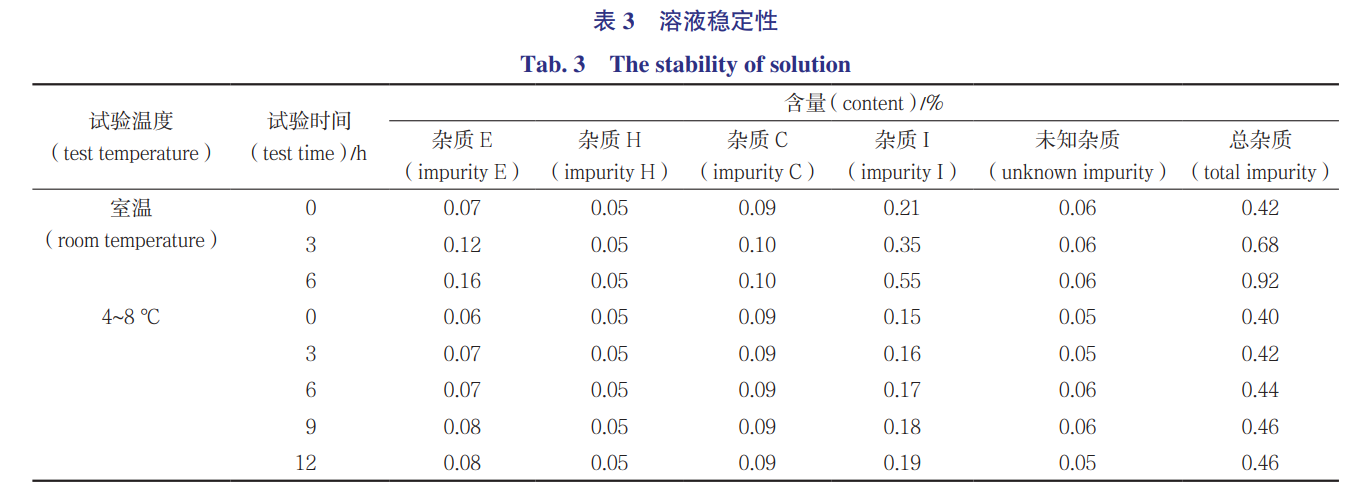
2.5溶液稳定性
取甲磺酸达比加群酯原料药10mg,精密称定,置10mL量瓶中,加30%乙腈溶解并稀释至刻度,摇匀,即得供试品溶液。该溶液在室温(25℃)下和4~8℃分别放置,并于0、3、6、12h分别进样测定,分别记录242、310、340nm波长下的色谱图。按照面积归一化法计算含量。除了杂质A~I按确定的检测波长进行检测,未知杂质(以达比加群为参比,相对保留时间为1.16)选择242、310、340nm3个波长中吸收最强的波长310nm计算含量。结果见表3。结果显示,供试品溶液在室温时,杂质E和杂质I增长明显,6h时已经增长翻倍,12h不再测定;在4~8℃时,12h内比较稳定。其他已知杂质均未检出。
2.6专属性试验
2.6.1空白溶剂取溶剂(30%乙腈)10µL,注入液相色谱仪,记录色谱图,结果显示溶剂对测定无干扰。
2.6.2强制降解取甲磺酸达比加群酯原料药10mg,分别进行强酸降解(1mol·L-1盐酸1mL,室温放置4h)、强碱降解(1mol·L-1氢氧化钠1mL,室温振摇0.5h)、氧化降解(3%双氧水,室温放置4h)、光照降解(4500lx日光灯光,照射24h)、高温降解(沸水加热1h),酸、碱降解必须进行中和,即得各降解样品。各降解样品用30%乙腈制成1mg·mL-1的降解样品溶液,同时制备相同浓度的未破坏样品的供试品溶液。取上述各溶液10µL,注入液相色谱仪,记录色谱图,见图3。在酸、碱降解条件下,主要降解得到杂质H,并且有一相对保留时间(以达比加群酯作参比)为0.9的主要降解杂质;在氧化、光照和高温降解条件下,均会降解得到杂质E;在光照和高温降解条件下,主要降解得到杂质E和杂质I。

2.7精密度和回收率试验
取甲磺酸达比加群酯原料药10mg,加30%乙腈5mL溶解,加入各杂质储备液适量,用30%乙腈稀释制成每1mL含杂质A~I及甲磺酸达比加群酯均约1μg的混合溶液,控温4~8℃。取上述混合溶液10μL连续进6次,记录色谱图。在选定的波长下测定各杂质,以各杂质的峰面积计算精密度试验的RSD。结果峰面积的RSD在0.19%~0.67%,满足精密度要求RSD≤2.0%,表明此方法精密度良好。精密称取甲磺酸达比加群酯原料药10mg,置10mL量瓶中,加5mL30%乙腈溶解,加入各杂质储备液适量,配制成各杂质质量浓度均分别为7.5、5和2.5μg·mL-1(相当于甲磺酸达比加群酯浓度的0.75%、0.5%、0.25%)的溶液各3份,作为回收率试验溶液,控温4~8℃,分别进样测定,计算得杂质A~I的回收率(n=9)分别为92.5%~94.5%[回收率平均值(n=3)分别为92.6%、94.2%、93.8%,RSD分别为0.14%、0.49%、0.53%]、93.4%~98.1%[回收率平均值(n=3)分别为95.4%、96.0%、94.7%,RSD分别为2.1%、2.5%、1.6%]、90.1%~97.2%[回收率平均值(n=3)分别为90.6%、95.9%、90.5%,RSD分别为0.64%、1.1%、0.67%]、92.8%~101.0%[回收率平均值(n=3)分别为94.5%、99.9%、99.3%,RSD分别为1.6%、1.1%、0.57%]、98.1%~100.7%[回收率品均值(n=3)分别为98.3%、100.4%、98.7%,RSD分别为0.21%、0.30%、0.21%]、99.8%~105.8%[回收率平均值(n=3)分别为100.1%、104.4%、105.6%,RSD分别为0.31%、0.37%、0.38%]、92.3%~97.5%[回收率平均值(n=3)分别为92.4%、94.4%、96.2%,RSD分别为0.14%、0.52%、1.2%]、95.9%~101.3%[回收率平均值(n=3)分别为96.5%、100.8%、98.1%,RSD分别为0.75%、0.50%、1.1%]和98.9%~103.8%[回收率平均值(n=3)分别为100.4%、102.6%、102.8%,RSD分别为1.7%、0.29%、1.6%],RSD分别为0.89%、1.9%、3.0%、2.8%、0.99%、2.3%、1.9%、2.1%和1.6%。
2.8样品测定
取3批甲磺酸达比加群酯原料药,分别按“2.5”项下方法制备供试品溶液;精密量取供试品溶液1mL,置100mL量瓶中,加30%乙腈稀释至刻度,摇匀,精密量取5mL,置10mL量瓶中,加30%乙腈稀释至刻度,摇匀,即得对照溶液。取供试品溶液、对照溶液各10μL,注入液相色谱仪,记录色谱图。已知杂质按照加校正因子的自身对照法来进行计算各杂质含量,未知杂质按照主成分的自身对照法进行计算(供试品中任何小于对照溶液主峰面积1/20的未知杂质峰可忽略不计),结果见表4。

制备方法[2]
甲磺酸达比加群酯的制备方法,其操作过程具体介绍如下:
(1)将3-[[[2-[[(4-脒基苯基)氨基]甲基]-1-甲基-1H-苯并咪唑-5-羰基](吡啶-2-基)氨基]-丙酸乙酯对甲苯磺酸盐、碳酸钾加入溶解剂中混合均匀;
具体用量方面,3-[[[2-[[(4-脒基苯基)氨基]甲基]-1-甲基-1H-苯并咪唑-5-羰基](吡啶-2-基)氨基]-丙酸乙酯对甲苯磺酸盐用量为40g,碳酸钾用量为56g;
混合操作时,所述溶解剂为丙酮-水体系;所述丙酮-水体系,具体由1000ml丙酮和550ml水混合均匀配制而成溶液。
所述3-[[[2-[[(4-脒基苯基)氨基]甲基]-1-甲基-1H-苯并咪唑-5-羰基](吡啶-2-基)氨基]-丙酸乙酯对甲苯磺酸盐,通过如下步骤制备获得:
A、将3-[[[2-[[(4-氰基苯基)氨基]甲基]-1-甲基-1H-苯并咪唑-5-基]羰基]吡啶-2-基氨基]丙酸乙酯、甲醇加入反应釜中;
具体用量方面,甲醇加入量为300g,3-[[[2-[[(4-氰基苯基)氨基]甲基]-1-甲基-1H-苯并咪唑-5-基]羰基]吡啶-2-基氨基]丙酸乙酯用量为50g,即,甲醇加入量为3-[[[2-[[(4-氰基苯基)氨基]甲基]-1-甲基-1H-苯并咪唑-5-基]羰基]吡啶-2-基氨基]丙酸乙酯质量的6倍;
B、通入氯化氢气体至饱和,室温反应24小时;
C、减压蒸干后加入甲醇300g溶解,通入氨气至饱和,室温反应24h;
D、然后加入对甲苯磺酸,搅拌析晶,过滤、洗涤(采用丙酮进行洗涤)、干燥,即得3-[[[2-[[(4-脒基苯基)氨基]甲基]-1-甲基-1H-苯并咪唑-5-羰基](吡啶-2-基)氨基]-丙酸乙酯对甲苯磺酸盐;
具体用量方面,对甲苯磺酸用量为31g;
最终制得3-[[[2-[[(4-脒基苯基)氨基]甲基]-1-甲基-1H-苯并咪唑-5-羰基](吡啶-2-基)氨基]-丙酸乙酯对甲苯磺酸盐42.0g。
(2)向步骤(1)中混合均匀的反应体系中加入氯甲酸正己酯,常温下(20℃左右)反应3h左右;然后4℃左右进行析晶,过滤得粗品;
对应步骤(1)中的用量,氯甲酸正己酯用量为11g。
(3)将步骤(2)所得粗品加精制溶剂乙醇300ml 于60℃左右溶解,溶解完全后,将反应液降温至4℃左右进行析晶,过滤,得达比加群酯25.0g。
(4)将步骤(3)所得达比加群酯20.0g加反应溶剂丙酮200ml 于42℃左右溶解,溶解完全后滴加甲磺酸3.2g,充分反应后将反应液降温至4℃左右析晶,过滤,将滤饼干燥7~8h,所得即为甲磺酸达比加群酯,共获得21.4g。
进一步对所获得成品进行检测,最终计算及检测结果表明:以3-[[[2-[[(4-脒基苯基)氨基]甲基]-1-甲基-1H-苯并咪唑-5-羰基](吡啶-2-基)氨基]-丙酸乙酯对甲苯磺酸盐计,重量收率为 66.9%;而高效液相色谱法(HPLC)测定纯度为:99.8%,单杂0.08%。
参考文献
[1]强文舟,张艳侠,傅强.HPLC法测定甲磺酸达比加群酯的有关物质[J].药物分析杂志,2020,40(04):672-680.
[2] [中国发明] CN201810729075.1 一种甲磺酸达比加群酯的制备方法


欢迎您浏览更多关于甲磺酸达比加群的相关新闻资讯信息