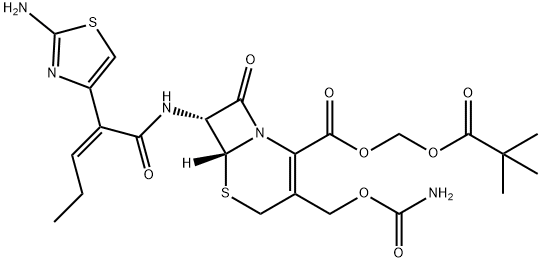
Description
Flomox was launched in Japan as an orally active cephalosporin for
respiratory and urinary tract infections, heptatic infections, ophthalmological and
otorhinolarynological infections, skinkoft tissue infections, and for use in gynacology,
dentistry and oral surgery. It can be prepared by condesation of 2(Z)-(2-(t-butoxycarbonylamino)
thiazol-4-yl)-2-pentenoic acid with 7-amino-3-(carbanoyloxymethyl)-
3-cephem-4-carboxylic acid pivaloyl methyl ester followed by deprotection.
Flomox is highly active against a wide variety of Gram-positive and Gram-negative
bacteria, except for several strains such as Pseudomonas aeruginosa and
enterococci, by acting as a cell wall synthesis inhibitor (β-lactamase stability against
TEM-1 type β-lactamases) and is more effective than cefaclor and cefdinir. Absorption
is improved by the pivaloyloxymethyl ester group which is easily lost by deesterification
during GI absorption to produce the biologically active form. The pivalic acid
generated quickly conjugates with carnitine and is excreted in the urine. The drop in
plasma levels of carnitine was dose dependent and returned to normal levels upon
termination of treatment.