ChemicalBook CAS数据库列表 决奈达隆
决奈达隆
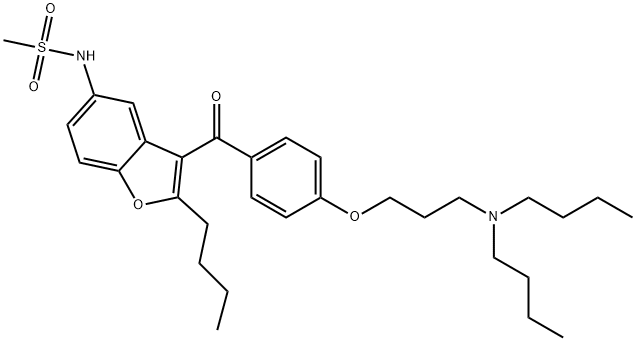
- CAS号:141626-36-0
- 英文名:Dronedarone
- 中文名:决奈达隆
- CBNumber:CB11117179
- 分子式:C31H44N2O5S
- 分子量:556.76
- MOL File:141626-36-0.mol
决奈达隆化学性质
- 熔点 :65.3°
- 沸点 :683.9±65.0 °C(Predicted)
- 密度 :1.143±0.06 g/cm3(Predicted)
- 储存条件 :2-8°C
- 溶解度 :≥27.84 mg/mL in DMSO; insoluble in H2O; ≥49.8 mg/mL in EtOH
- 形态 :solid
- 酸度系数(pKa) :7.40±0.30(Predicted)
- 颜色 :White to off-white
- CAS 数据库 :141626-36-0(CAS DataBase Reference)
安全信息
决奈达隆性质、用途与生产工艺
-
抗心律失常新药
心房纤颤(AF) 是一种有着潜在生命威胁的疾病,由于人口老龄化,全球心房纤颤发病率,死亡率不断走高,并成为新的公共卫生难题。美国的心房纤颤患者人数达 250 万人,欧盟的患者人数达 450万人。据统计,我国30岁以上人群,房颤患病率为0.77%,随着年龄而增加,男性高于女性(0.9%∶0.7%)。引起房颤的疾病主要包括高血压、冠心病、瓣膜性心脏病、自发性扩张型心肌病、心力衰竭。房颤患者的主要症状为不规则的心室率,控制心率在症状的急性处理和维持治疗中占一定地位。胺碘酮在控制心率、恢复窦性心律方面作用良好,但其结构中的碘成分相关的靶器官的不良反应限制了其临床应用,诸如对肺、甲状腺和肝脏的损害,上述不良反应与其结构中的碘关系密切。通过对胺碘酮结构修饰得到新的抗心律失常药决奈达隆(Dronedarone)。决奈达隆 (Dronedarone),具有与胺碘酮类似的电生理特性,但结构中不含碘,因此可以避免碘所引起的严重的心脏外的副作用。
决奈达隆于2009年7月被美国食品与药物管理局(FDA)批准上市,2009年在欧盟批准上市,也是10年来首个在欧盟获准的抗心律失常新药。决奈达隆即N-[2-丁基-3-[4-[3-(二丁氨基)丙氧基]苯基]-5-苯并呋喃基]-甲烷磺酰胺,它是为了减少非心脏不良反应而通过对胺碘酮进行结构修饰得到的苯并呋喃的衍生物。其去掉了胺碘酮中的碘原子,以减少甲状腺及其他器官毒性;在苯并呋喃一侧增加了甲磺酰基,降低了亲脂性,从而缩短药物的半衰期,减少药物的组织蓄积作用和可能的神经毒性。2011年美国版和2010年欧洲版心房颤动指南均推荐决奈达隆用于心房颤动的治疗。决奈达隆治疗各种不伴有严重 AF 患者的心室率和维持窦性速率方面安全有效,可以成为替代胺碘酮有效药物,决奈达隆同时可以降低 AF 患者甚至是持久 AF 患者的死亡率 , 因此有望成为不伴有严重心衰的 AF 患者一线用药。且中-高危 AF 病人使用抗凝剂对决奈达隆的使用有益。 -
药理作用
决奈达隆作为Ⅲ类抗心律失常药物,对多种离子通道具有抑制作用。研究表明,决奈达隆10μmol·L-1可以明显降低犬心室肌细胞快速激活的延迟整流钾通道电流( 97%,P< 0.05 )和 L 型钙通道电流 ( 76.5%,P < 0.05)。同样剂量的决奈达隆可以缩短蒲肯野纤维的动作电位时程(action potential dutation,APD)。同时,决奈达隆可以降低豚鼠心室肌细胞动作电位的最大上升速度( dV·dt-1max ),且该效应强度与决奈达隆浓度成正比关系。另一研究表明,决奈达隆可延长兔心肌细胞的RR、QT间期,心室 APD50、APD90分别延长20 %及49%,且同等剂量决奈达隆的上述效果强于胺碘酮。同时,在心室细胞复极过程中,决奈达隆可阻断人类电压门控钾离子通道基因( hERG) 亚单位编码的复极钾电流,而这种被阻断的电流可能和长 QT 综合 征及尖端扭转型室速有关,而该电流在( 37 ± 1 ) ℃可被决奈达隆阻断。人类心房细胞研究发现,应用决奈达隆0.3μmol ·L-1时抑制 I Na 达23 %,而在 3μ mol ·L-1时抑制 I N a达 97%,呈明显剂量依赖关系。对急性心肌梗死大鼠的研究提示,决奈达隆在防止急性心肌梗死后出现的快速及缓慢型心律失常方面与胺碘酮有相似的效果。这些实验结果可能是决奈达隆抗房性心律失常的机制之一。
综上,决奈达隆可抑制心肌细胞的多种离子通道,延长APD 时相,但对不同通道抑制的程度不同。进一步研究证实,决奈达隆降低冠脉灌注压不是通过 抑制环氧化酶(如吲哚美辛)发挥作用,而是决奈达隆造成了左旋精氨酸 (L-NO ARG ; 3- 100AM)的浓度反应曲线的右移 ,而这种效应和决奈达隆剂量相关。上述实验证实决奈达隆和胺碘酮类似,存在扩张冠状动脉作用。
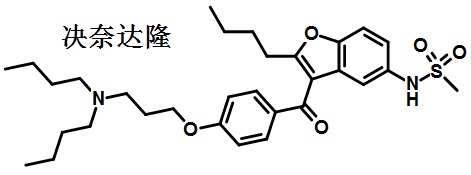
图1为决奈达隆的结构式 -
药代动力学
决奈达隆为苯并呋喃衍生物,化学结构类似胺碘酮,但其化学结构与胺碘酮比较发生了2个改变:去除了碘基;增加甲烷磺酰胺基。因此不但消除了碘基对甲状腺激素等的影响,甲烷磺酰胺基也减小了决奈达隆的亲脂性,减少了组织积聚,不必给予负荷量时即能较快达到治疗浓度,缩短了半衰期,减少药物的组织蓄积。决奈达隆血浆半衰期为1~2d,而胺碘酮的半衰期为6~8 周。
吸收:因为体循环前首过代谢,无食物时决奈达隆的绝对生物利用度是低,约4%。当决奈达隆与高脂肪餐给药增加至约15%,在食物条件下口服后3至6小时内达到血浆决奈达隆和主要循环活性代谢物(N-debutyl代谢物)峰浓度。每天2次重复给予400mg,在治疗4至8天内达到稳态和决奈达隆的平均积蓄比范围从2.6至4.5。主要N-debutyl代谢物的稳态Cmax和暴露与母体化合物相似,决奈达隆及其N-debutyl代谢物的药代动力学均中度偏离剂量正比例:剂量增加2-倍导致约2.5-至3.0倍增加Cmax和AUC。
分布:血浆蛋白结合率 98%,主要与清白蛋白结合,体内分布广泛,静脉给药后,表观分布容积可达到1200 ~1400 L。一日两次口服给药,5~7 d达稳态血药浓度。
代谢:决奈达隆被广泛代谢,主要被CYP3A。初始通路包括N-debutylation形成形成活性N-debutyl代谢物,氧化脱氨作用形成无活性的丙酸代谢物,和直接氧化作用。各种代谢物进行进一步代谢产生超过30种未鉴定的代谢物,N-debutyl 谢物表现出药效学活性但是强度为决奈达隆的1/10至1/3。
排泄:用口服决奈达隆质量平衡研究中(14C-标记)约6%的标记剂量在尿中被排泄,主要为代谢物(尿中无未变化化合物排泄),而84%在粪中排泄,主要为代谢物。决奈达隆及其N-debutyl活性代谢物至少占血浆中由此产生的放射性小于15%。静脉给药后决奈达隆的血浆清除率范围从130至150L/h。决奈达隆的消除半衰期范围从13至19小时。 -
药物相互作用
决奈达隆主要经肝脏经细胞色素氧化酶450(CYP450)广泛代谢,既是CYP4503A的底物,也是CYOP4503A和CYP4502D的中度抑制剂,同时又是P-糖蛋白的底物,可抑制药物的P-糖蛋白转运,因此,经CYP4503A和CYP4502D代谢P-糖蛋白转运的药物均可能与决奈达隆发生相互作用。
1.CYP 3A 酶强抑制剂(抗真菌药和大环内酯类) 可以使其血浆药物浓度升高 25 倍,因此应减少决奈达隆剂量或禁止合用 ;
2. CYP3A 酶中等抑制剂如钙通道阻滞剂可以使其血浆药物浓度升高1.4 ~1.7 倍,因此应减少钙拮抗剂剂量;
3.肝药酶CYP450诱导剂利福平、苯巴比妥、卡马西平、苯妥英钠可使该药的血浆药物浓度降低 5 倍,因此应增加决奈达隆剂量;
4.决奈达隆( 400mg,bid) 与CYP3A底物辛伐他丁同服,可使后者血药浓度增高 2~4倍,因此合用时应密切监测肌毒;
5.与P-糖蛋白转运地高辛合用可使其浓度增高 1.7~2.5 倍,因此地高辛剂量减半;
6.与CPY4503A底物西罗莫司他克莫司合用可能增加西罗莫司他克莫司血浓度,因此应密切监测西罗莫司他克莫司血浓度;
7.与CYP2D底物β-受体阻滞剂阻滞剂入美托洛尔合用可使其浓度升高 1 . 3~1. 6 倍,因此合用时应适当调整上述药物的剂量,以免引起毒性;
8.与CPY2C底物华法林合用,可使华法林血浓度升高,INR从2.0~3.0升至4.8,因此合用时应减少华法林剂量。 -
适应症
决奈达隆是抗心律失常药物,适用于阵发性或持续性心房颤动(AF)或心房扑动(AFL)患者,减低住院风险,近期AF/AFL发作和伴心血管风险因子患者,窦性心律或心律可复律的患者。
有关决奈达隆的药理作用、药代动力学、药物相互作用、适应症、不良反应等是由Chemicalbook的旭艳编辑整理。(2016-03-25) -
不良反应
1.新心衰或心衰恶化;
2.钾消耗利尿药的低钾血症和低镁血症;
3.QT延长;
4.在AF或AFL患者每天2次400mg决奈达隆的安全性评价是基于5项安慰剂对照研究,ATHENA,EURIDIS,ADONIS,ERATO和DAFNE。这些研究中,总共6285例患者被随机化和治疗,3282例患者用MULTAQ 400mg每天2次,和2875例用安慰剂。跨研究平均暴露为12个月。ATHENA最长随访为30个月;
5.在临床试验中,因为不良反应过早停药,决奈达隆-治疗患者发生11.8%和安慰剂-治疗组7.7%。用MULTAQ治疗停药的最常见原因是胃肠道疾患(3.2 %相比安慰剂组)和QT延长(1.5%相比安慰剂组0.5%);
6.在5项研究用MULTAQ每天2次400 mg观察到最频不良反应为腹泻、恶心、腹痛、呕吐和虚弱; -
禁忌
1.心衰类别IV或最近失代偿症状性心衰;
2.II-或III-度房室(AV)阻断或病态窦房结综合征(除使用功能性心脏起搏器);
3.心动过缓<50 bpm;
4.同时用强CYP3A抑制剂;
5.同时用延长QT间隔及可诱发尖端扭转型室性心动过速(Torsade de Pointes)药物和草药;
6.QTc Bazett间隔≥500 ms;
7.严重肝损伤;
8.妊娠;
9.哺乳母亲。
决奈达隆上下游产品信息
上游原料
下游产品
决奈达隆 试剂级价格
- 更新日期:2025/02/08
- 产品编号:HY-A0016
- 产品名称:决奈达隆 Dronedarone
- CAS编号:141626-36-0
- 包装:5 mg
- 价格:300元
- 更新日期:2025/02/08
- 产品编号:HY-A0016
- 产品名称:决奈达隆 Dronedarone
- CAS编号:141626-36-0
- 包装:10mM * 1mLin DMSO
- 价格:367元
决奈达隆生产厂家
- 公司名称:Xunteng International Trading Co.,Limited
- 联系电话:
- 电子邮件:
- 国家:中国香港
- 产品数:878
- 优势度:55
141626-36-0, 决奈达隆相关搜索: