Arsphenamine - Drug Application, Reach Process and Discovery History
In 1858 David Livingstone, the Scottish medical missionary who explored much of Central Africa, recommended Fowler’s Solution for the alleviation of the symptoms of sleeping sickness. This was to take on greater significance after David Bruce demonstrated in 1896 that trypanosomes were present in the blood of animals afflicted with nagana, a form of sleeping sickness, and could be temporarily eliminated by administering Fowler’s Solution. Trypanosomes were protozoa somewhat larger than red blood corpuscles. Several varieties were subsequently shown to be responsible for what had previously been classified as 80 different tropical diseases affecting both men and animals. The most devastating of these in humans was sleeping sickness, caused by Trypanosoma brucei infection carried by bloodsucking tse-tse flies. In one recorded instance, the mortality had risen from 13 to 73% between the years 1896 and 1900. There were even fears that the disease could depopulate the whole of Central Africa, where it was being transmitted by the tse-tse fly, especially after an epidemic in Uganda killed 250 000 people in the first two decades of the twentieth century. Attempts to prepare vaccines proved futile.
In 1899, Alfred Lingard at Muktesar in India tried unsuccessfully to cure horses infected with the form of trypanosomiasis known as ‘surra’ by administering Fowler’s Solution. Three years later, Alphonse Laveran and Felix Mesnil at the Pasteur Institute announced that they had been able to infect laboratory mice and rats with two varieties of the causative organism of trypanosomiasis. Subcutaneous injections of the sodium salt of arsenious acid into the infected rodents resulted in a rapid disappearance of trypanosomes from their blood, but the parasites reappeared within a few days to cause the death of the animals.
Antoine Be´champ synthesised the sodium salt of the meta-anilide of phenylarsonic acid in 1863. At the beginning of the twentieth century it was marketed by the Vereinigte Chemische Werke in Charlottenburg with the unsubstantiated claim that it was 40–50 times less toxic than any of the inorganic arsenicals previously used. On this basis it was given the overly optimistic name of Atoxyl1.28 The director of the factory at Charlottenburg, Ludwig Darmsta¨ dter, had been particularly interested in finding a cure for cancer and had sponsored some of Paul Ehrlich’s work in this field at the Institute for Experimental Therapy in Frankfurt. Whether Darmsta¨ dter sent Ehrlich a sample of Atoxyl1 with a view to its being evaluated in cancer or in trypanosomiasis is not clear. Atoxyl1 was, however, the first substance Ehrlich investigated after receiving cultures of trypanosomes from Edmond Nocard of the Pasteur Institute. Unfortunately, it proved to be inactive.29 After this, Ehrlich began his investigation of chemotherapeutic dyes, which preoccupied him until 1905, when he was surprised to read a paper by Wolferstan Thomas, of the Liverpool School of Hygiene and Tropical Medicine, describing the success of Atoxyl1 in the treatment of animals experimentally infected with trypanosomiasis.30 Ehrlich at once reopened his investigation of Atoxyl1 and confirmed the validity of the results obtained by Thomas. He then realised that his earlier study had been misleading because it had been carried out on isolated cultures of trypanosomes, rather than on infected animals. This implied that Atoxyl1 either stimulated immunity or else had to be metabolically converted to an active form by the animal before any trypanocidal effect could be exerted. Meantime, the bacteriologist Robert Koch was asked by the German Sleeping Sickness Commission to evaluate Atoxyl1 in East Africa, where the mortality from the disease was reaching alarming proportions. Koch established that a 500 mg injection of the drug could cause the disappearance of trypanosomes from the blood for up to eight hours. His recommendation was that effective treatment would require constant medication for six months, a protocol that would present the risk of blindness through damage to the optic nerve in up to 2% of patients. This convinced Ehrlich that it would be worth his while examining analogues of Atoxyl1 to see whether the ratio of efficacy to toxicity could be improved. Thanks to the generosity of the widow of Frankfurt banker George Speyer, who had donated a million marks, and also to John D. Rockefeller, who supported Ehrlich’s research, a chemotherapy institute was built alongside the existing Institute for Experimental Therapy. Called the Georg Speyer-Haus, it was officially opened in September 1906. There were excellent facilities for chemists to synthesise the analogues of Atoxyl1 required by Ehrlich.
Ehrlich was fond of coining Latin aphorisms to express the complex ideas underlying his researches. To describe the principle guiding the design of the new chemotherapeutic agents he proposed to synthesise, he used the phrase corpora non agunt nisi fixata. This was his way of explaining that infecting organisms are not killed unless the chemotherapeutic agent has a high affinity for them. Ehrlich termed such drugs parasitotropic. Their toxic effects he attributed to their also being, to varying extents, organotropic. His ideal agent would have a high therapeutic index because it exhibited a high parasitotropic activity and a low organotropic activity. The therapeutic index could be measured simply by comparing both the curative and lethal doses of the test compounds in mice. Around the time he moved into the Georg Speyer-Haus, he began to accept the ideas on the existence of drug receptors newly propounded by the Cambridge physiologist John Langley. Ehrlich then advanced the hypothesis that the parasitotropic action of arsenicals was due to their binding to arsenoceptors on the surface of the parasites, these being specific types of chemoceptors. It was his fervent hope that he could develop drugs that, like the antibodies discovered by his former colleague Emil von Behring, would act like ‘magic bullets’ insofar as they would bind only to receptors in the parasite and not the patient.
Initially, Ehrlich believed the scope for molecular modification would be limited since Atoxyl1 was known to be the chemically unreactive anilide of arsenic acid. His unexpected discovery that the drug reacted with nitrous acid to form a diazonium salt led him to the realisation that the structure originally assigned to it by Be´champ was incorrect. Ehrlich correctly concluded that Atoxyl1 was the sodium salt of 4-aminophenylarsonic acid, which meant that methods existed for preparing a wide range of its analogues.

In the spring of 1907, Ehrlich signed a formal contract with the nearby Cassella Dye Works, granting them exclusive rights to the commercial exploitation of his arsenic compounds in return for their sponsorship of his research. The arrangement continued after the Hoechst Dyeworks took control of Cassella in 1908. Ehrlich’s chief chemist, Alfred Bertheim, then synthesised a range of Atoxyl1 derivatives with substituents on the amino group. One of these was the N-acetyl derivative of Atoxyl1, known as arsacetin. It was less toxic to mice, but when administered at the high dose level required to effect a cure it caused the mice to rotate in the manner of Japanese waltzing mice. This indicated damage to the vestibular nerve (associated with balance) similar to that produced by Atoxyl1, and it suggested that arsacetin would likewise cause blindness. Recalling his earlier work on lead poisoning, Ehrlich became convinced that this nerve damage was due to chronic arsenic poisoning caused by the large amount of arsenic that had been administered. In seeking a more potent series of arsenicals he hoped to avoid this particular problem. To this end chloro, hydroxy, cyano, sulfonic acid and amino groups were introduced into the benzene ring, but all exhibited their own particular types of toxicity over and above neurotoxicity. None of them exhibited any enhancement of potency.
The discovery that higher concentrations of phenylarsonic acids were required to kill test tube cultures of trypanosomes than could possibly be achieved when curing infected mice led Ehrlich to conclude that they were undergoing metabolic activation, presumably through a process of chemical reduction. To test this hypothesis, he asked Bertheim to prepare the two possible types of reduction products from phenylarsonic acids. One type, the arsenoxides, were highly toxic to trypanosomes, but their general toxicity to the tissues of the host was also high. They were also very irritant when injected, due to the presence of impurities. The second series, the arsenobenzenes, were not as potent, although still more potent than the phenylarsonic acids, and they were less toxic to the host. This meant that small doses could be given to avoid neurotoxicity from chronic arsenic poisoning. Ehrlich subsequently restricted his investigations to arsenobenzenes, unaware that the neurotoxicity of his phenylarsonic acids was mainly due to impurities.
Ehrlich and his contemporaries believed that the arsenobenzenes consisted of two fragments linked by a double bond between their respective arsenic atoms. It was only years later that this was shown to be incorrect by Kraft and his colleagues in Russia, who established that the arsenobenzenes are polymeric mixtures formed from several molecules joined by single bonds between their arsenic atoms. Polymers cannot penetrate mammalian cells, which explains why the toxicity of arsenobenzenes was much weaker than that of the arsenoxides formed from only two molecules joined via arsenic–oxygen–arsenic linkages. The chemotherapeutic action of both arsenobenzenes and arsenoxides was shown in the 1920s to be due to the release of arsenite molecules which had only a single arsenic atom that reacted with thiol groups in receptors on the surface of the parasites.
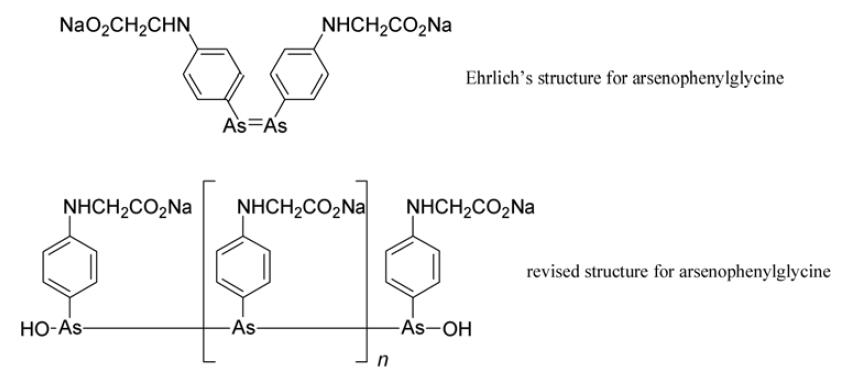
Arsenophenylglycine (compound 418) emerged as a promising trypanocidal agent. It was administered to patients in 1907 and for many it proved safe and effective. Unfortunately, in a small number severe and often fatal hypersensitivity to it was exhibited. Despite this, some physicians deemed it acceptable to continue to use it for those forms of trypanosomiasis with a high mortality rate. Ehrlich strongly disagreed.
Insertion of a hydroxyl group on the 4-position of the benzene ring was readily achieved by warming diazotised Atoxyl1 in water. Reduction of the product yielded arsenophenol. This was highly effective against trypanosomes, but was prone to oxidation to the severely irritant arsenoxide and exceedingly difficult to purify. However, Ehrlich’s prior experience with chemotherapeutic dyes (see below) convinced him that the introduction of a substituent adjacent to a phenolic hydroxyl group enhanced chemotherapeutic activity. An arsenophenol substituted in this manner was synthesised by Bertheim in 1907 as potential trypanocidal agent 606. The assistant who tested it told Ehrlich that it was ineffective, and consequently it remained on a shelf in the laboratory for over a year before being re-examined.
The isolation of the organism that caused syphilis, Treponema pallidum, was reported from the Reichsgesundheitsamt in Berlin by Fritz Schaudinn and Erich Hoffmann in 1905. The following year, Hoffmann visited the Georg Speyer-Haus and told Ehrlich that this spirochaete was in many ways similar to trypanosomes. He asked for samples of Ehrlich’s new compounds for testing on syphilitic patients in his clinic at Bonn, where he had just been appointed to the chair of dermatology. Ehrlich agreed, urging great care when using the arsenicals. Shortly after this, Uhlenhuth found that Atoxyl1 had a curative effect against syphilis in chickens, but was inferior to mercurials and could cause blindness through optic nerve damage. Ehrlich, meantime, arranged for his friend Albert Neisser, director of the dermatology clinic at Breslau, to take arsacetin and arsenophenylglycine to Java, where he had managed to inoculate apes with T. pallidum. The results were published in 1908 and confirmed the efficacy of arsenophenylglycine.34 This was the only way in which Ehrlich was then able to have his compounds tested on animals. The arrangement lasted until the spring of 1909, when Ehrlich was joined by Sacachiro Hata who had developed a method of infecting rabbits with syphilis while working in the Kitasato Institute in Tokyo. On his arrival, Hata was asked to test every arsenical that had been synthesised by Bertheim and his colleagues over the preceding three years. Working swiftly and with precision, he discovered that compound 606 had outstanding curative properties in the rabbits infected with syphilis! As a result, Ehrlich arranged for the Farbwerke Hoechst to apply for a patent on ‘606’; the application was submitted on 10 June 1909. Further details of the synthesis were published later.
Disappointment over the hypersensitivity reactions among patients receiving arsenophenylglycine led Ehrlich to exercise great caution over the assessment of ‘606’. Once the results of animal studies confirmed that it was likely to be safe and effective, samples of ‘606’ were sent to Iversen at the Obuchow Hospital for Men in St Petersburg and Alt at Uchtspringe (Altmark). Iversen was the first to administer the drug to patients when he successfully treated relapsing fever, a disease transmitted by lice. The causative organism, Borrelia recurrentis, was similar to the T. pallidum. Alt, on the other hand, spent three months evaluating the safety of ‘606’ in dogs before he would let two of his assistants volunteer to be injected with the new arsenical. Only after this was the drug administered to several syphilitic patients suffering from general paralysis of the insane. Once its relative safety had thus been confirmed, samples of ‘606’ were given to Schreiber at Magdeburg, where trials were initiated on large numbers of patients with primary syphilis. By January 1910, several other trusted investigators had received ‘606’ for trials. Ehrlich insisted on personally checking the records of every patient who received the drug, and it was not until the following April that he was prepared to make any public announcement about ‘606’.
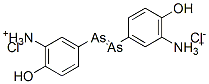
On 19 April 1910, Ehrlich told the Congress for Internal Medicine at Weisbaden of his work leading up to the discovery of ‘606’. Hata then described his experiments and Schreiber explained how he had been able to cure syphilis with ‘606’. These announcements received enthusiastic applause from those present, and in the following days the press throughout the world carried headline reports of the new drug that could cure syphilis. The immediate result of this publicity was that the Georg Speyer-Haus was beseiged by physicians and patients anxious to obtain samples of ‘606’. Letters also poured in from around the world. Under this unprecedented pressure, Ehrlich modified his policy of supplying the drug only to a small circle of trusted colleagues. He agreed to provide the physicians, at no charge, with a small number of vials of the drug that had been prepared under his supervision, on the condition that he received full reports of every case treated. Between April and December 1910, some 65 000 vials of ‘606’ were freely supplied in this manner while the necessary plant for its commercial production was being installed in the Farbwerke Hoechst. Production began there at the end of the year. ‘606’ was then marketed under the proprietary name of Salvarsan1, the approved name given to it later being arsphenamine. To the general public it remained known as either ‘606’ or ‘Ehrlich–Hata 606’.
Many syphilitic patients were cured by a single intravenous 900mg dose of arsphenamine. This injection was prepared from powdered arsphenamine hydrochloride in a sealed ampoule. Prior to being injected, this had to be treated with the correct amount of dilute alkali to form a solution containing the highly unstable sodium salt, a procedure that brought many complaints from physicians. As more experience was gained, Ehrlich realised repeated dosing was necessary.38 Typically, 600mg doses were repeated every few days until a total of 5 g of arsphenamine had been administered. Four to six weeks later, this was repeated and then another one or two similar courses of treatment were required. In all, each patient would receive 25–30 injections.
Despite early setbacks mainly because of careless administration, arsphenamine analogues remained the standard treatment for syphilis until the end of the Second World War, when adequate supplies of penicillin became available. There can be no question whatsoever that arsphenamine was the first major chemotherapeutic agent and that Ehrlich rightly deserves to be remembered as the founder of modern chemotherapy.
Shortly after starting to produce arsphenamine, the Hoechst Dyeworks patented neoarsphenamine (Neosalvarsan1), a water-soluble derivative that was made less sensitive to oxidation by condensing some of the amino groups on the polymer with sodium sulfoxylate, an antioxidant widely used in the dyestuffs industry.40 Trials revealed this to be less potent than arsphenamine. This worried Ehrlich, but he was reluctantly persuaded that it should be made available because of the preference of the medical profession for a preparation that required only the addition of water to the ampoule immediately before injection. Neoarsphenamine ultimately superseded arsphenamine, usually being co-administered with either mercury ointment or oral mercury. It also became the standard treatment for anthrax until the introduction of antibiotics.
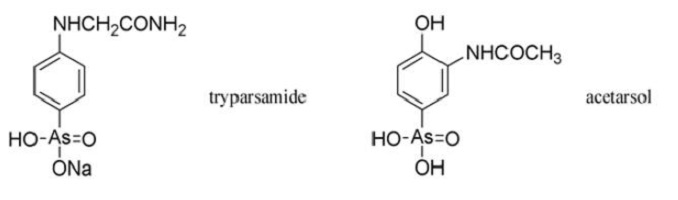
The Rockefeller Foundation patented tryparsamide, but they issued licenses free of charge to manufacturers who wished to produce it. It was truly fitting that the Rockefeller Institute, founded in 1901 with an endowment of $200 000, should succeed in fulfilling Ehrlich’s original objective only four years after his death. John D. Rockefeller had been one of the first backers of Ehrlich’s work on arsenicals.
Ernest Fourneau at the Pasteur Institute questioned Ehrlich’s belief that the neurotoxicity of phenylarsonic acids was due to the large doses required. Fourneau was convinced that the blindness caused by Atoxyl1 was due to impurities, and this led him to examine thousands of phenylarsonic acids, from which acetarsol emerged as a valuable antisyphilitic and amoebicide. Acetarsol had been used in 1911 as an intermediate in the preparation of some of Ehrlich’s arsphenamine analogues. Fourneau arranged for it to be manufactured by Poulenc Freres under the proprietary name of Stovarsol1, a pun on his own name (French: fourneau=stove).
Fears that Lewisite1 would be used by the enemy during the Second World War led the British Ministry of Supply to set up a team of scientists charged with the responsibility of finding an antidote. Based in the department of biochemistry at Oxford and led by Rudolf Peters, they established that arsenite reacted with two adjacent thiol groups on the vital pyruvate oxidase enzyme system. The discovery that this reaction could be competitively inhibited by the presence of simple sacrificial molecules that also contained two adjacent thiol groups ultimately resulted in the preparation of a highly effective antidote, namely dimercaprol (BAL, or British Anti-Lewisite).49,50 It was a viscous liquid that could be applied to the skin in an ointment, or else be injected. This wartime episode constitutes one of the finest examples of rational drug design, and dimercaprol is still used as an antidote to poisoning by arsenic, mercury and other metallic poisons that react with thiol groups in the pyruvate oxidase system.