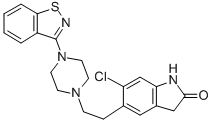
Both p.o. and i.m. formulations of ziprasidone were launched in Sweden for the
treatment of schizophrenia and agitated psychoses. It is the sixth marketed atypical
antipsychotic after clozapine, risperidone, olanzapine, sertindole and quetiapine. The
synthesis of ziprasidone involves a novel one-step process for the preparation of 3-(1-
piperazinyl)-1,2-benzisothiazole followed by coupling with a chlorooxindole fragment.
Ziprasidone is a very potent 5-HT2A/D2 antagonist with a ratio of about 11 in favor of the
serotonin receptor. It also shows very high 5-HT2c antagonistic activity, high 5-HT1A
agonistic and 5-HT1D antagonistic activity, as well as moderate antagonism of α1 and H1
receptors and moderate norepinephrine and serotonin reuptake inhibition. Its complex
binding profile for serotonin and dopamine receptors resulted during clinical trials in high
antipsychotic efficacy with low extrapyramidal side effects and also in antidepressive
action with low propensity for weight gain in opposition to other atypical and typical
neuroleptics. An intramuscular formulation of ziprasidone was demonstrated to be superior
to haloperidol, a conventional neuroleptic, for the short-term treatment of agitation in
acutely psychotic patients. When administered orally in the fed state, this well-tolerated
agent which strongly binds to plasma proteins shows a bioavailability of about 60% which
is almost 2 fold greater than in the fasted state. It is transformed into 4 circulating major
metabolites by different enzyme systems. The small QTc prolongation observed with
ziprasidone was found to be comparable to other antipsychotic drugs and it is considered
to be without significant risk.