Triheptanoin Protects Motor Neurons
There is increasing evidence that energy metabolism is disturbed in Amyotrophic Lateral Sclerosis (ALS) patients and animal models. Treatment with triheptanoin, the triglyceride of heptanoate, is a promising approach to provide alternative fuel to improve oxidative phosphorylation and aid ATP generation. Heptanoate can be metabolized to propionyl-CoA, which after carboxylation can produce succinyl-CoA and thereby re-fill the tricarboxylic acid (TCA) cycle (anaplerosis).
Anaplerosis and Triheptanoin as a Treatment Approach
Anaplerosis serves to “re-fill” the levels of C4 carbon-deficient TCA cycle intermediates to improve energy supply during periods of increased energy need [53]. Anaplerotic enzymes include pyruvate carboxylase (Pcx), which produces oxaloacetate and glutamic pyruvic transaminases 1 and 2 (Gpt1 and 2), which catalyze the reaction pyruvate + glutamate < = > α-ketoglutarate + alanine (Fig 1).
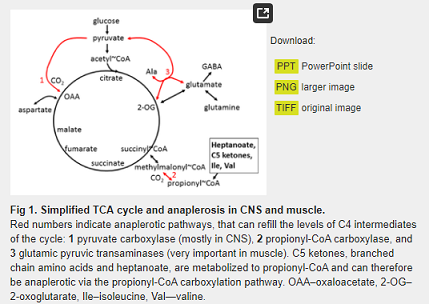
Triheptanoin, the triglyceride of heptanoic (C7) acid, not only provides an alternative fuel source in the form of medium chain fats, that are typically absent in a standard normal diet, but it can also be anaplerotic. It is a novel metabolic therapeutic that is being used in clinical pilot studies to treat patients with various disorders associated with metabolic dysfunction. This includes genetic metabolic disorders of fatty acid oxidation [53,54] and neurological and muscular disorders [55–57]. Triheptanoin provides the body with heptanoate, which diffuses into the mitochondria of cells in both the CNS and peripheral tissues to be metabolized to acetyl-CoA and propionyl-CoA by β-oxidation. Also, the liver converts heptanoate into the “C5 ketones”, β-hydroxypentanoate and β-ketopentanoate, which are then released into blood. After being taken up via monocarboxylate transporters into cells of various tissues, including the CNS, “C5 ketones” are also β-oxidized to acetyl-CoA and propionyl-CoA. Carboxylation of propionyl-CoA produces succinyl-CoA, a C4 TCA cycle intermediate via propionyl-CoA carboxylation, an anaplerotic pathway that has been described in various tissues [58–60] (Fig 1). An adequate supply of C4 TCA cycle intermediates is important for optimal oxidation of fuels by the TCA cycle [53,61].
There is now increasing evidence that triheptanoin improves energy metabolism in the CNS and muscle of patients and in animal models of different diseases, including brain and muscle in Huntington’s Disease patients [55,62] and models of epilepsy [60,63]. Triheptanoin also was found to provide alternative fuel to the brains of patients and mice with glucose transporter 1 deficiency, which show impaired glucose uptake into the CNS [56,59]. In addition, there is evidence that triheptanoin can prevent cell loss in the brain in mouse models of stroke [64] and Canavan disease [65] by improving mitochondrial respiration.
Despite the beneficial effects observed following triheptanoin treatment in multiple models of disease and neurodegeneration, the use of triheptanoin as a potential therapeutic in ALS remains unexplored. Thus, we used hSOD1G93A mice to test the hypotheses that 1) triheptanoin attenuates motor neuron death and delays the onset of motor symptoms, and that 2) the expression of enzymes involved in energy metabolism is diminished in muscle and can be rescued by triheptanoin.
Materials and Methods
Animals
All experiments were approved by the University of Queensland Animal Ethics Committee and followed the guidelines of the Queensland Animal Care and Protection Act 2001. Wild-type and hSOD1G93A mice (B6.Cg-Tg(SOD1*G93A)1Gur/J, stock no. 004435, Jackson laboratory, Bar Harbor, ME, USA), were generated by mating hSOD1G93A males with C57BL/6 wild-type females (University of Queensland). Mice were housed in a 12 hour light, 12 hour dark cycle, and had free access to food and water. Experimenters were blinded to animal genotype (until the mice started expressing the ALS phenotype) and treatment.
Triheptanoin Treatment
Starting at P35, female wild-type and hSOD1G93A mice were given either control or triheptanoin-containing diet treatment until they were sacrificed. Untreated mice received SF11-027, a diet with ingredients typical to other mouse diets (Specialty Feeds, Glen Forrest, WA, AUS). Treated wild-type and hSOD1G93A mice received a matched formulation (SF11-028, Specialty Feeds) in which 35% of the calories were from triheptanoin. This dose was chosen, as it is also used in the clinic to treat patients with metabolic disorders and has shown beneficial metabolic and protective effects in several animal models of various disorders [55,56,60,62–64,66]. The diets were matched in protein, mineral, antioxidant and vitamin content relative to their caloric densities [66]. Triheptanoin replaced sucrose, some of the complex carbohydrates and long chain fats. For the motor neuron count experiments, triheptanoin was provided by Ultragenyx Pharmaceutical Inc. (Novato, CA, USA). All other experiments were performed with triheptanoin obtained from Sasol GmbH (Germany).
Motor Neuron Counts
Mice were deeply anesthetized with pentobarbital (120 mg/Kg i.p. Provet, Northgate, QLD, AUS) and euthanized by decapitation. Spinal cords were flushed out of the spinal cavity using a cold phosphate buffered saline (PBS) filled syringe that was fitted with a blunted 23-gauge needle. Spinal cords were immediately fixed in 4% paraformaldehyde (PFA, pH 7.4) for 24 to 48 hours and then embedded in liquid paraffin. Serial, transverse sections (16μm) were obtained using a Leica Rotary Microtome. Motor neurons were identified after staining with thionine (0.1%) in acetate buffer (pH = 3.9). The L4 to L5 regions were identified according to standard anatomical guidelines described in [67] using an Olympus BX61 upright light microscope with 10x, 20x and 40x objectives. Briefly, the L4 and L5 sections were identified according to the area of the gracile fasciculus and dorsal corticospinal tract relative to the spinal cord central canal. In L3 sections, the dorsal corticospinal tract is smaller than in L4 and L5. Motor neuron numbers in L4 and L5 were determined using stereological principles based on the counting of every 10th section with a total of 11 to 16 sections counted [68]. Every tenth consecutive L4 and L5 spinal cord section was counted until the sacral dorsal commissural nucleus appeared concomitant with a decrease in size of the gracile fasciculus and dorsal corticospinal tract, which are markers for L6. Motor neurons were identified by a large, darkly stained cell body, a pale nucleus with a continuous boundary and one or more darkly stained nucleoli [67].
Blood ketone and glucose measurements
Blood plasma was collected in EDTA-coated tubes from the mice used for motor neuron counting, centrifuged at 2,000g for 10 mins. It was then stored at -80°C until analysis. Plasma levels of the ketone β-hydroxybutyrate and glucose were determined using Cayman colorimetric assay kits (Ann Arbor, MI, USA), according to the manufacturer’s instructions.
Behavioral Testing and Observation
Animals underwent behavioral testing approximately 3–4 hours into the light cycle. All behavioral testing was conducted in an environment with minimal stimuli so as to minimize any possible effects caused by changes in external stimuli. Animals were weighed before every test session. Mice were observed, and disease progression tracked and graded according to a neurological score sheet [69] to ensure that any non-ALS related deaths were excluded from the study. In accordance with ethical requirements, hSOD1G93A mice that became too weak to reach the food hoppers were provided with wet chow on the floor of the cages. The endpoint of the study was defined as the mouse being unable to right itself within 15 seconds after being placed on its back. Upon reaching end-stage or 25 weeks of age, transgenic mice and their respective wild-type littermates were euthanized with pentobarbital (120 mg/kg, i.p., Provet). Tissues, including the gastrocnemius muscle, were collected for subsequent analysis. To measure the time point when body weight loss started, we defined the day where a loss of more than 10% in an individual mouse occurred relative to its mean body weight from week 12 to 17. Also all subsequent three body weight measurements were ≤ 90% of the original mean weight.
Hind Limb Grip Strength Test
Hind limb grip strength tests were conducted twice a week using a T-bar force transducer (Ugo Basile, Varese, ITA). The animal was held by the tail, ensuring its hind limbs were gripping the T-bar before being pulled downwards at a 60° angle. The reading on the force transducer was taken only if both hind limbs released the bar at the same time. The average of 10 trials per mouse was recorded for each training session [70]. To compare time points of grip strength loss, we determined the time point where a grip strength loss of more than 30% occurred in an inidividual mouse relative to its mean grip strength of week 9 to 13 and the subsequent three measurements were ≤ 70% of the original average strength.
Rotarod Test
Rotarod tests were conducted with 10 repeats once a week using a rotarod designed for mice (Ugo Basile). Animals were placed on the rod, which was then rotated for 3 min at 25 revolutions per minute [70]. The time at which the animal fell off was recorded. We defined the age of balance loss on the rotarod when this time was zero.
RNA Extraction, cDNA Synthesis, Quantitative Real Time PCR Assay
After euthanasia, the gastrocnemius muscle was quickly removed and frozen in liquid nitrogen. To extract RNA, muscle samples were pulverized in liquid nitrogen with a cold mortar and pestle, dissolved with TRI reagent (Life Technologies, Carlsbad, CA, USA) and extracted according to the manufacturer’s instructions. Contaminating DNA was removed by DNAse I treatment and cDNA was synthesized using the Tetro cDNA synthesis kit (Bioline, London, UK) according to the manufacturer’s instructions.
Results
Triheptanoin Protected Against Motor Neuron Loss in hSOD1G93A Mice
A defining hallmark of ALS is progressive motor neuron loss. Here, we confirmed that relative to wild-type mice, 38% of lumbar L4-L5 motor neurons were lost by 70 days of age in hSOD1G93A mice (Fig 2, two-way ANOVA p<0.0001 for genotype, Bonferroni multiple comparisons post hoc test p<0.0001, n = 7–10 per group). To determine if triheptanoin can protect motor neurons, we initiated triheptanoin treatment at P35 in female hSOD1G93A mice at 35% of caloric intake until P70. This dose was chosen, as it is also used in the clinic to treat patients with metabolic disorders and has shown beneficial metabolic and protective effects in several animal models of various disorders [55,56,60,62–64,66]. Starting triheptanoin treatment at P35 in hSOD1G93A mice resulted in higher motor neuron numbers by 18% relative to untreated hSOD1G93A mice (two-way ANOVA p = 0.0126 for treatment, Bonferroni multiple comparisons post hoc test p<0.05), amounting to a 33% protection against motor neuron loss.
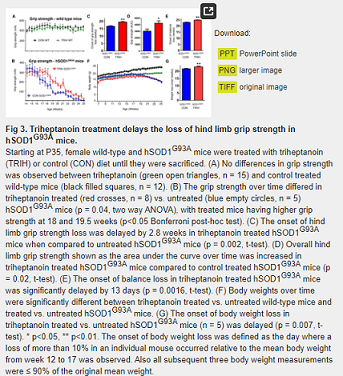
We investigated the extent to which triheptanoin delays the onset of the progressive loss of muscle strength in hSOD1G93A mice. Hind limb grip strength tests were used to assess the course of disease progression in treated vs. untreated mice. There was no observable difference between the mean hind limb grip strength of the treated (n = 15) vs. untreated (n = 12) wild-type mice. Mean hind limb grip strength for both groups of wild-type mice consistently fell between 300 and 600 mN (Fig 3A). The hind limb grip strength of hSOD1G93A mice on both treatments never exceeded 400 mN (Fig 3B). The time at which reduced hind limb grip strength became apparent was significantly later in triheptanoin treated hSOD1G93A mice when compared to untreated hSOD1G93A mice (n = 8 for triheptanoin treated, n = 5 for untreated mice; Fig 3B, p = 0.04, two way ANOVA). Bonferroni’s multiple comparisons tests indicated that at 18 and 19.5 weeks of age, triheptanoin treated hSOD1G93A mice had higher hind limb grip strength when compared to untreated hSOD1G93A mice (Fig 3B, p<0.05). Untreated hSOD1G93A mice began to lose hind limb grip strength at 16.7 weeks of age, but the time of onset of the loss of hind limb grip strength was delayed by 2.8 weeks in hSOD1G93A mice that received oral triheptanoin (p = 0.002, Fig 3C). The area under the curve for the grip strength over time for each mouse treated with triheptanoin was increased by 38% (p = 0.024, t-test, Fig 3D).
Related articles And Qustion
See also

Lastest Price from TRIHEPTANOIN manufacturers

US $0.10-10.00/KG2025-08-15
- CAS:
- 620-67-7
- Min. Order:
- 0.10000000149011612KG
- Purity:
- 98%
- Supply Ability:
- 200TONS

US $0.00-0.00/kg2025-08-15
- CAS:
- 620-67-7
- Min. Order:
- 1kg
- Purity:
- 99%
- Supply Ability:
- 2000tons