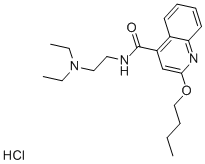
The synthetic methods of Dibucaine hydrochloride
Dibucaine hydrochloride blocks the generation and conduction of nerve impulses by reducing the permeability of neuronal cell membranes to sodium ions. This action reversibly stabilizes the cell membrane and inhibits depolarization, resulting in failure of action potential propagation and subsequent conduction block. Dibucaine is recognized as one of the most potent, most toxic, and longest-duration local anesthetics.[1] The synthesis method of dibucaine is as follows.
Synthetic method 1[2]

Fig.1. Synthetic method 1
Toluene (120 kg) was charged into a dry and clean 500 L reactor, followed by the addition of 2-butoxyquinoline-4-carboxylic acid (24.5 kg) under stirring; after homogenization, thionyl chloride (14.3 kg) was added, and the mixture was gradually heated to 60-70°C; after the reaction was complete, excess thionyl chloride was distilled off, yielding a toluene solution of 2-butoxyquinoline-4-carbonyl chloride for subsequent use.
To a 10 wt% sodium hydroxide solution (48 kg) was added N,N-Diethylethylenediamine (13.9 kg) with stirring until uniform, the solution was cooled to below 0°C, and then the prepared 2-butoxyquinoline-4-carbonyl chloride toluene was added dropwise at -5 to 0°C; after the reaction, the mixture was warmed to 20-30°C, acidified to pH 2.0-3.0 with 30% hydrochloric acid, and stirred for 30 min; the stirring was stopped and the mixture was allowed to stand for 10 min; phase separation was performed, the organic phase (toluene) was treated for reuse; to the lower aqueous phase (cinchocaine hydrochloride solution) was added activated carbon (0.5 kg), and the mixture was stirred at 30-40°C for 30 min for decolorization, then filtered; the filtrate was adjusted to pH 10.0-11.0 with 30% sodium hydroxide solution and stirred for 30 min; then the stirring was stopped and the mixture was allowed to stand for 10 min; phase separation was conducted, and the organic phase was retained. To the organic phase was added isopropan (120 kg) with stirring until uniform; at 30-40°C, the pH was adjusted to 3.0-4.0 with 36% hydrochloric acid, and the mixture was stirred for 30 min; then cooled to -5-0℃ and crystallized for 3 h, followed by centrifugation; the wet product was dried to afford dibucaine hydrochloride (31.2 kg) in 82.1% molar yield with 99.94% HPLC purity.
Synthetic method 2[3]
Step 1: 2-Hydroxyquinoline-4-carboxylic acid and toluene were charged into a reactor, followed by dropwise addition of thionyl chloride; after reaction completion, thionyl chloride was removed by rotary evaporation, and then N,N-Diethylethylenediamine was added dropwise with stirring at 20-75°C for 1-24 h; the mixture was filtered, washed with water, and dried to yield 2-chloro-N-[2-(diethylamino)ethyl]quinoline-4-carboxamide.
Step 2: The 2-chloro-N-[2-(diethylamino)ethyl]quinoline-4-carboxamide was added to n-butanol and sodium n-butoxide, heated under reflux with stirring; after reaction completion, toluene was added for extraction and crystallization to afford purified dibucaine. The purified dibucaine was dissolved in a solvent, followed by dropwise addition of ethanolic hydrochloric acid; the precipitated solid was stirred for 2 h, filtered, and dried to give dibucaine hydrochloride.
Synthetic method 3[4][5]

Fig.2. Synthetic method 3
Step 1: 2-Hydroxyquinoline-4-carboxylic acid was added to toluene at room temperature, and thionyl chloride was added dropwise under stirring; after reaction completion, thionyl chloride was removed under reduced pressure to afford intermediate 2-chloroquinoline-4-carbonyl chloride.
Step 2: The 2-chloroquinoline-4-carbonyl chloride was dissolved in toluene, and N,N-diethylethylenediamine was added to the solution with heating; after reaction, the mixture was washed with water, crystallized, filtered, and dried to yield intermediate 2-chloro-N-[2-(diethylamino)ethyl]quinoline-4-carboxamide.
Step 3: The resulting 2-chloro-N-[2-(diethylamino)ethyl]quinoline-4-carboxamide was added to a solution of n-butanol and sodium n-butoxide at room temperature and refluxed; after completion, the mixture was cooled, water was added with stirring, and the phases were separated with the aqueous phase discarded; the organic phase was dried over anhydrous sodium sulfate, filtered, and concentrated under reduced pressure; the concentrate was treated to obtain a processing solution, which was crystallized overnight at low temperature to give crude cinchocaine; the crude product was dissolved in solvent at elevated temperature, maintained for a period, filtered while hot, and the filtrate was crystallized by freezing, filtered, and dried to afford purified dibucaine. The purified dibucaine was added to acetone, dissolved with stirring at elevated temperature, and hydrochloric acid in acetone solution was added dropwise; after maintaining temperature with stirring for 0.5 h, the mixture was cooled for crystallization, filtered, and dried to yield purified dibucaine hydrochloride.
References
[1] Kuroda, Yoshihiro, Locations of local anesthetic dibucaine in model membranes and the interaction between dibucaine and a Na+ channel inactivation gate peptide as studied by 2H- and 1H-NMR spectroscopies, [J] Biophysical Journal (1996), 71(3), 1191-1207.
[2]Shandong Chenghui Shuangda Pharmaceutical Co., Ltd., Production process for preparing Cinchocaine hydrochloride by one-pot method, [P] CN110981801
[3]Hefei Yuanzhi Pharmaceutical R & D Co., Ltd., Preparation method of dibucaine hydrochloride, [P] CN107586277
[4] Kunming Yuanrui Pharmaceutical Co., Ltd., Method for preparation of Cinchocaine hydrochloride, [P] CN106496120
[5] Chiesi Farmaceutici S.p.A., Compound with antiinflammatory activity, process for preparation thereof and pharmaceutical compositions therefrom. [P] EP123146


Lastest Price from Dibucaine hydrochloride manufacturers

US $0.00-0.00/kg2025-04-21
- CAS:
- 61-12-1
- Min. Order:
- 1kg
- Purity:
- 0.99
- Supply Ability:
- 800kg

US $0.00-0.00/kg2025-04-16
- CAS:
- 61-12-1
- Min. Order:
- 1kg
- Purity:
- 99%
- Supply Ability:
- 500