The synthesis method of Upadacitinib hemihydrate
Overview
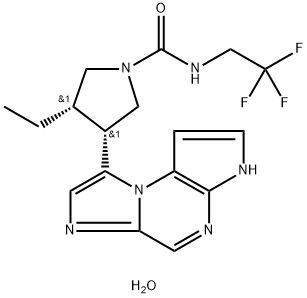
Upadacitinib, a once-daily orally administered selective JAK1 inhibitor developed by AbbVie, was approved for the treatment of moderate to severe RA. Similar to tofacitinib and baricitinib, upadacitinib received a USFDA-boxed safety warning due to increased risks of thrombosis. The selective inhibition of cytokine JAK1-dependent inflammatory pathways associated with RA (e.g., interleukin-6 and interferon-γ) minimizes off-target physiological side effects associated with JAK2 and JAK3 engagement.
Synthesis method
The synthesis of fragment 191 began with a Larock indole synthesis employing dibromopyrazine 186[1]. This facile reaction afforded indole 188 in good yield. Tosylate protection followed by palladium-catalyzed Buchwald−Hartwig amination provided ethyl carbamate 191.

The chiral pyrrolidine fragment 198 was rapidly synthesized starting from an in situ Michael addition−Dieckmann condensation of Cbz-protected glycine ethyl ester 192 and ethyl acrylate (193) in the presence of NaOt-Bu to afford 194. Reaction with trflic anhydride gave enol triflate 195, which underwent a Suzuki−Miyaura reaction with ethylboronic acid to provide 4-ethylpyrrolidine 196 in good yield. Acid hydrolysis set the stage for a ruthenium-catalyzed asymmetric hydrogenation using (S)-Segphos as a chiral phosphine ligand. The synthetic sequence to the sulfoxonium ylide was carried out sequentially without the isolation of intermediates. This sequence began with saponification of ethyl ester 196, setting the stage for a ruthenium-catalyzed asymmetric hydrogenation using ruthenium diacetate and (S)- Segphos as a chiral phosphine ligand.
With the chiral acid in hand, activation with CDI and displacement with the methylide (formed from deprotonation of dimethylsulfonyl chloride) afforded sulfoxonium 198. Treatment of the sulfoxonium with lithium bromide under acidic conditions gave the α-bromo ketone, which was carried on to the next step. Displacement of the bromide with carbamate 191 furnished intermediate 199, the ring-closure precursor, in good yield. Cyclization/dehydration under mild conditions followed by basic detosylation afforded tricycle 200 in 92% yield. Removal of the carboxybenzoyl group with 10% palladium hydroxide on carbon gave the free amine, which was isolated as bis(hydrochloride) salt 201. The reaction of 2,2,2- trifluoroethylamine with CDI and slow addition of imidazolide 201 provided upadacitinib hemihydrate in 87% yield.
References
[1] Andrew C. Flick. “Synthetic Approaches to the New Drugs Approved during 2019.” Journal of Medicinal Chemistry 64 7 (2021): 3604–3657.
You may like
See also



US $0.00-0.00/g2025-06-17
- CAS:
- 2050057-56-0
- Min. Order:
- 1g
- Purity:
- 99%
- Supply Ability:
- 20kg

US $0.00/kg2025-06-13
- CAS:
- 2050057-56-0
- Min. Order:
- 1kg
- Purity:
- 98%
- Supply Ability:
- 1000kg