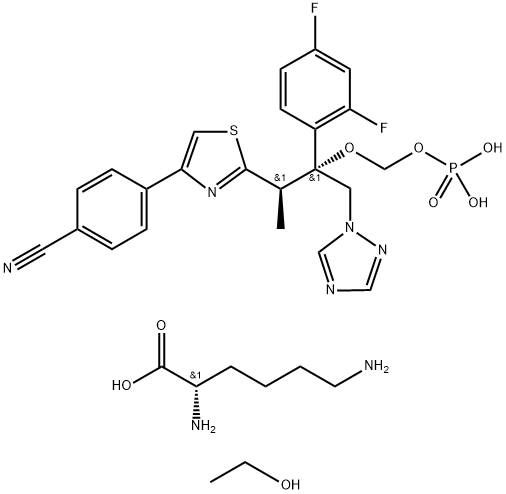
The preparation methods of Ravuconazole and its prodrug F-RVCZ.
Description
Ravuconazole is a novel triazole antifungal agent exerting broad and potent antifungal activity. Fosravuconazole L-lysine ethanolate (F-RVCZ) is an orally administered, broad-spectrum antifungal drug approved in Japan for treating onychomycosis in 2018. This drug is a prodrug of Ravuconazole with improved solubility and oral bioavailability[1]. The following describes the preparation methods of Ravuconazole and its prodrug F-RVCZ.Z.
Biological function
Initially discovered by Eisai, Ravuconazole was licensed to Bristol-Myers Squibb (BMS) for worldwide development, excluding Japan, in 1996. However, BMS terminated the development of the drug in 2004, and Eisai reacquired the worldwide development, manufacturing, and marketing rights. Like other azole drugs, the antifungal activities of Ravuconazole derive from the inhibition of ergosterol biosynthesis and block the 14α-demethylation pathway present in many strains of yeasts and molds. The lowering of ergosterol levels leads to the accumulation of 14α-methyl sterols impairs normal structure and functions of cell membranes, ultimately resulting in growth inhibition or death of fungal cells. F-RCVZ exhibited higher efficacy (higher initial cure rates and lower recurrence rates), an improved safety profile (lower hepatic functional disorders), and an improved dosing regimen (once daily for 12 weeks) over existing standards of care such as terbinafine and itraconazole.
The mechanism underlying the antifungal activities of RVCZ is considered to involve inhibition of ergosterol biosynthesis, as is the case with other azole antifungals, and this activity is potently exerted against a broad spectrum of dermatophytes and pathogenic fungi, including the genera Trichophyton, Candida, Aspergillus, and Cryptococcus.
Preparation methods
In addition to several disclosures describing the gram-scale synthesis of ravuconazole and related precursors, a robust plant-scale preparation has been described by researchers at BMS (shown below)—this route utilized lactate as a starting material for the preparation of arylpropanone. First, methyl ester was converted to a morpholine amide in the presence of catalytic sodium methoxide. The alcohol was subsequently protected to generate tetrahydropyranyl ether. Realtime infrared reaction monitoring allowed for the safe formation of Grignard reagent from the corresponding bromide, which was then reacted with amide to furnish aryl ketone after aqueous acetic acid quench. Corey−Chaykovsky epoxidation and subsequent epoxide opening were performed in a single-step, telescoped process. Once epoxidation was complete, heating the reaction mixture to 90 °C triggered a triazole-mediated epoxide-opening sequence to form alcohol. The stereochemical outcome of the epoxide-forming step is dictated by the adjacent chiral center, provided in 8.6:1 dr. Removal of the tetrahydropyranyl protecting group generated an intermediate diol, which was converted to trisubstituted epoxide via selective mesylation of the secondary alcohol. Generation of lithium cyanide in situ from acetone cyanohydrin and LHMDS followed by subsequent addition to epoxide delivered the α-cyano alcohol in 90% yield, which was subsequently converted to thioamide monohydrate salt by treatment with diethyl dithiophosphate and sulfuric acid. Condensation of the thioamide with 2-bromo-4′-cyanoacetophenone in hot ethanol resulted in thiazole formation, completing the preparation of Ravuconazole.
-2-(2,4-difluorophenyl)-1-Methyl-2-[(phosphonooxy)Methoxy]-3-(1H-1,2,4-triazol-1-yl)propyl]-4-thiazolyl]benzonitrile and ethanol (1:1:1) (9CI)")
The conversion of Ravuconazole to the highly water-soluble prodrug fosravuconazole L-lysine ethanolate (II) has been described by the scientists at Eisai, as shown below. First, Ravuconazole was O-alkylated with di-tert-butyl chloromethylphosphate to furnish phosphate ester. The subjection of ester to trifluoroacetic acid (TFA) and aqueous sodium hydroxide provided the free acid, which was subsequently converted to fosravuconazole L-lysine ethanolate (II)[2].
-2-(2,4-difluorophenyl)-1-Methyl-2-[(phosphonooxy)Methoxy]-3-(1H-1,2,4-triazol-1-yl)propyl]-4-thiazolyl]benzonitrile and ethanol (1:1:1) (9CI)")
References
[1] Shinichi Watanabe, Akihiro Okubo, Ichiro Tsubouchi. “Efficacy and safety of fosravuconazole L-lysine ethanolate, a novel oral triazole antifungal agent, for the treatment of onychomycosis: A multicenter, double-blind, randomized phase III study.” Journal of Dermatology 45 10 (2018): 1151–1159.
[2] Andrew C. Flick. “Synthetic Approaches to New Drugs Approved during 2018.” Journal of Medicinal Chemistry 63 19 (2020): 10652–10704.