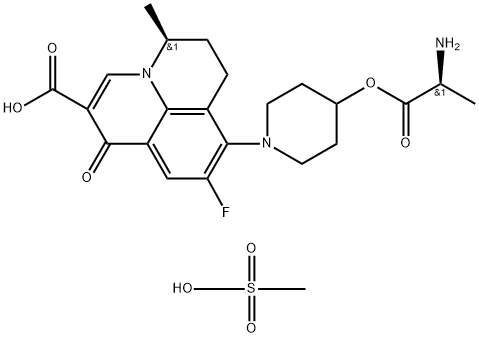
Synthesis of Alalevonadifloxacin mesylate
Synthesis of Alalevonadifloxacin mesylate
Alalevonadifloxacin mesylate is synthesized using 2-Bromo-4,5-difluoroacetylaniline as raw material through a series of chemical reactions. The specific synthesis method is as follows:

2-Bromo-4,5-difluoroacetylaniline was treated with crotonaldehyde under Skraup-Doeber-Von Miller conditions to generate quinoline 2 in 67% yield. Catalytic hydrogenation using palladium on carbon reduced the carbon− bromine bond within 2. Filtration of the reaction mixture to remove the catalyst followed by the addition of platinum on carbon and resubjection to hydrogenation conditions gave tetrahydroquinoline 3 in 97% yield.
Compound 3 was treated with 2,3-di-O-benzoyl-L-tartaric acid (L-DBTA) followed by recrystallization from 60% aqueous methanol to give S-isomer 4 in 35% yield and 100% ee. Although not shown, the recovered Risomer from the resolution could be racemized by treatment with methanesulfonic acid (MsOH). Compound 4 was treated with diethylethoxymethylenemalonate (EMME) in polyphosphoric acid (PPA) and treated with HCl to give tricyclic acid 5 in 88% yield. Chelation of 5 with boron triacetate (generated in situ) gave the cyclic borate complex 6, which was further reacted with 4-hydroxypiperidine to give levonadifloxacin 8 in good yield for the 2 steps. Coupling 8 with Boc-alanine followed by removal of the Boc group and salt formation with methanesulfonic acid gave alalevonadifloxacin mesylate in 95% yield.