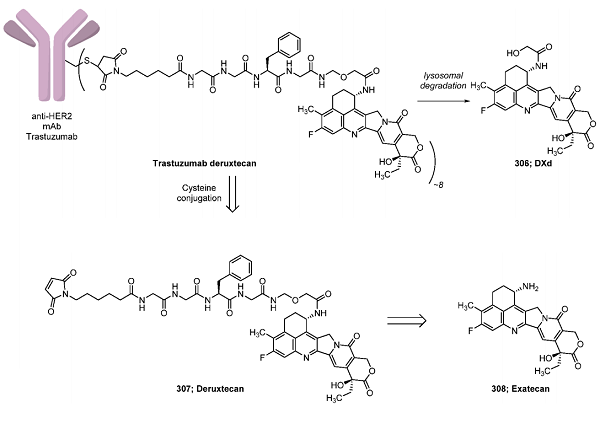
How to synthesize Trastuzumab Deruxtecan?
Synthesis of Exatecan Precursor 315

The synthesis of deruxtecan makes use of a late-stage coupling of exatecan and advanced linker intermediate 325. The synthesis of exatecan began with bromination of 2-fluoro-1- methyl-4-nitrobenzene (309) with NBS/sulfuric acid in heptane, making way for subsequent nitro reduction and acetylation of the resulting aniline to provide compound 310. The three-step sequence was completed in 37% overall yield, providing 310 as a white solid after a series of aqueous workup events and crystallization. Coupling of this bromide with 3-butenoic acid (311) under Heck conditions (Pd(OAc)2/P(o-tol)3) and subsequent hydrogenation of the resulting mixture of alkene isomers resulted in intermediate 312 after precipitation from heptane. Activation of the resulting carboxylic acid in 312 with TFA/TFAA initiated an intramolecular cyclization to provide tetrahydronapthalenone 313 after precipitation from a cold solution of acetonitrile/water. A THF solution of this carbocycle was sequentially subjected to nitration conditions followed by heterogeneous reduction, ultimately providing bis(amino acetate) 314 after workup and isolation. Selective aniline deacetylation with hot aqueous HCl provided intermediate 315 after precipitation and recrystallization from acetone.
Synthesis of Exatecan (308)

The remaining synthetic steps to exatecan are shown above. The subjection of 315 to advanced intermediate 316 provided the core structure of exatecan via a PPTS-mediated Friedlaender-type reaction. This reaction occurred under refluxing conditions in o-cresol and toluene and relied first on the condensation of the ketone within 316 and the aniline within 315. The reaction of these two intermediates gave rise to intermediate enamine 317, which then underwent dehydrative intramolecular cyclization to generate quinoline 318 as a mixture of diastereomers on a 170 g scale. In this regard, treatment of the mixture of 318 with aqueous methanesulfonic acid under refluxing conditions in ethylcyclohexene and 2-methoxyethanol resulted in the formation of the dihydromesylate amine salt of exatecan as a mixture with epi-exatecan salt 319, which can be separated by recrystallization. After the separation of the isomers, the undesired isomer 319 could also be recycled and converted to exatecan via an amine reprotection and deprotection sequence, which was made possible by isomerization of 319 under the deprotection reaction conditions. Using this overall process, the desired mesylate salt dihydrate of exatecan (308) was isolated after a series of aqueous washes and recrystallization events.
Synthesis of Advanced Deruxtecan Linker Intermediate 325

An improved synthetic route to linker intermediate 325 on a multikilogram scale that enabled the isolation of deruxtecan in crystal form without the need for chiral chromatography was also recently reported by Daiichi Sankyo. The subjection of glycine acid 320 to a previously developed oxidative decarboxylation process with Pb(OAc)4 and acetic acid in refluxing THF provided aminal 321 after precipitation from cyclopentyl methyl ether. One-pot acetyl cleavage and subsequent two-carbon homologation with benzyl glycolate under Lewis acid conditions provided benzyl ester 322. Removal of Fmoc within 322 followed by coupling with dipeptide 323 (prepared by coupling of L-phenylalanine and N-9-fluorenylmethoxycarbonylglycylglycine ) enabled the formation of compound 324 over the one-pot, two-step sequence after crystallization of the product from 2- propanol. Cleavage of the benzyl ester from 324 was performed under heterogeneous conditions with H2 catalyzed by a palladium/carbon−ethylenediamine complex, providing 325 following filtration and precipitation from ethyl acetate.
Synthesis of Deruxtecan (307)
With exatecan 308 and linker 325 in hand, the final steps to deruxtecan were conducted by joining these fragments under peptide coupling conditions with EDCI and HOBt·H2O at room temperature, providing intermediate 326. Isolation of the product in acceptable purity was possible via treatment of the organic layer from the reaction with activated carbon and MgSO4 and subsequent crystallization from THF. Finally, Fmoc deprotection of 326 with DBU in THF and the reaction of the resulting amine with 6-maleimidocaproic acid (327) generated deruxtecan (307) on a 400 g scale. By using this method, precipitation of the product from acetone and 1-propanol yielded 191 g of 307, which could also be prepared in various other crystal forms suitable for conjugation to the trastuzumab antibody.
Conjugative Endgame for Trastuzumab Deruxtecan

Preparation of the final drug product, trastuzumab deruxtecan, necessitated conjugation of deruxtecan to the anti-HER2 monoclonal antibody trastuzumab, generating the ADC with a loading of approximately eight deruxtecan units per antibody. This conjugation proceeded through the treatment of a PBS/EDTA buffer solution of the trastuzumab antibody with TCEP and 1 M dipotassium hydrogen phosphate, providing a final pH of 7.4. Incubation at 37 °C led to a reduction of the intrachain disulfide bonds, giving rise to intermediate 328. Subjection of this mixture to excess deruxtecan (9.2 equiv per antibody) followed by reoxidation and quenching of any nonreacted sulfide bonds with aqueous N-acetylcysteine provided trastuzumab deruxtecan in ∼8:1 DAR after purification by gel filtration.
Reference
[1] Andrew C. Flick. “Synthetic Approaches to the New Drugs Approved during 2019.” Journal of Medicinal Chemistry 64 7 (2021): 3604–3657.