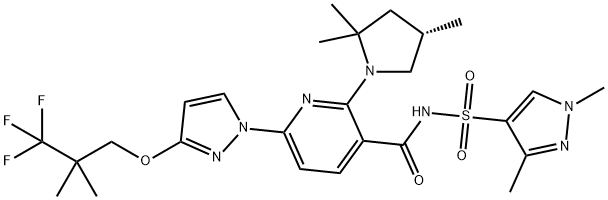
How to synthesize Elexacaftor?
Description
Trikafta, the combination of elexacaftor (VX-445), tezacaftor (VX-661) and ivacaftor (VX-770), was approved for therapy of cystic fibrosis (CF) patients with at least one allele of the CFTR mutation F508del. Ivacaftor is a cystic fibrosis transmembrane conductance regulator (CFTR) potentiator, while elexacaftor and tezacaftor are CFTR correctors. Mutations in the genes encoding CFTR proteins affect chloride and bicarbonate transport across the apical membranes of epithelial cells, leading to mucosal buildup and, ultimately, chronic infections and organ dysfunction. Utilizing the two classes of CFTR modulators in combination serves to simultaneously increase defective CFTR protein processing and trafficking and improve CFTR protein activity at the cell surface[1].
Synthesis method
Two synthetic routes to elexacaftor were disclosed by Vertex in 2018 and 2019. A review summarizing these routes was also recently published. Vertex prepared elexacaftor through a convergent approach in their first-generation strategy, synthesizing three fragments coupled sequentially about a lynchpin dichloronicotinic acid core. Pyrazole fragment 354 originated from methyl (E)-3-methoxyacrylate (351). This Michael acceptor underwent an addition−elimination reaction when treated with hydrazine monohydrate, followed by cyclization and Boc protection to afford pyrazolone 352 in 71% yield. A Mitsunobu reaction involving trifluoroalcohol 353 and subsequent Boc deprotection with HCl provided pyrazole 354.

Two methods were disclosed to synthesize chiral pyrrolidine 358, a five-step enzymatic resolution procedure and a four-step ring-contraction route. The contraction approach was described on 257 kg scale and was originally published by BF Goodrich in 1980. Piperidone 355 was treated with base in chloroform in the presence of a phase-transfer reagent, which triggered a fascinating mechanistic sequence. This sequence is described in detail in the literature. It relies on epoxidation, piperidine ring cleavage via nitrogen-assisted opening of the dichloroepoxide intermediate, subsequent imine hydrolysis, and recyclization to furnish the ring-contracted product. Following this, the chiral methyl stereocenter could be installed through asymmetric hydrogenation using rhodium or ruthenium catalysis, providing high yield and enantioselectivity. A final LiAlH4 reduction and salt formation afforded pyrrolidine 358 in 75% yield over two steps[2].

To complete the synthesis of elexacaftor, carboxylic acid 359 was protected as the tert-butyl ester. This was followed by a substitution reaction with pyrazole 354 to yield 366 in 95% over the two steps. Ester 366 was hydrolyzed under acidic conditions in 91% yield. Activation of the acid with CDI and exposure to sulfonamide 368 gave the penultimate chloropyridine 369 a 93% yield. A final substitution of the remaining pyridyl chloride with hindered pyrrolidine 358 afforded elexacaftor in 90% yield.
References
[1] Guido Veit , Gergely L. Lukacs, Christian Vaccarin . “Elexacaftor co-potentiates the activity of F508del and gating mutants of CFTR.” Journal of Cystic Fibrosis 20 5 (2021): Pages 895-898.
[2] Andrew C. Flick. “Synthetic Approaches to the New Drugs Approved during 2019.” Journal of Medicinal Chemistry 64 7 (2021): 3604–3657.