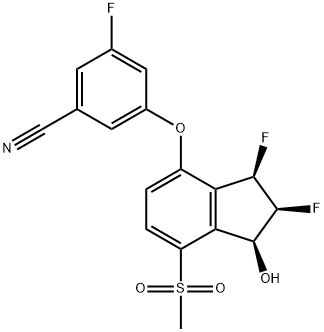
How is Belzutifan synthesised?
Synthesis of Belzutifan
Belzutifan was prepared by a synthetic route starting from 3,4-dihydrocoumarin, followed sequentially by the synthesis of Indanone Core and Indanone Ketal, and finally concentrating on three consecutive stereocentres present on the indane ring. The specific synthesis steps are as follows:
Step 1: Synthesis of Indanone Core 33.8
The optimized route starts with 3,4-dihydrocoumarin (33.1), which can be regioselectively brominated with NBS under Lewis acid conditions to generate intermediate 33.2 in high yield (91%), making way for a tandem lactone ring-opening/SNAr reaction. This was successfully achieved by subjecting 33.2 to K2CO3 in NMP and H2O, leading to the desired ring-opening product, which spontaneously participated in an SNAr reaction in the presence of 3,5-difluorobenzonitrile (33.3) and 18-crown-6 to generate intermediate 33.5. In this case, the addition of 18-crown-6 was found to be essential for promoting the SNAr reaction while minimizing the formation of bis-SNAr addition products. After extraction with i-PrOAc, the crude material was reacted with oxalyl chloride in MTBE/DMF, providing the corresponding acyl chloride 33.6, which underwent an intramolecular Friedel−Crafts reaction in the presence of AlCl3. This sequence generated indanone 33.7 in 75% overall yield from brominated intermediate 33.2. Methyl sulfone installation was achieved under copper-mediated conditions using sodium methanesulfinate to generate 33.8 in 78% isolated yield.

Step 2: Synthesis of Indanone Ketal 33.13
Starting from indanone 33.8, protection of the ketone moiety as its corresponding ketal intermediate297 33.9 preceded development of a continuous-flow light-mediated benzylic bromination step. This course was pursued to help prevent overbromination and deketalization byproducts observed under thermal bromination conditions. As such, intermediate 33.11 could be generated by radical bromination with 1,3-dibromo-5,5-dimethylhydantoin (33.10) in acetonitrile in the presence of citric acid and visible light, generating the crude reaction mixture, which was carefully quenched with 2,6- lutidine and 1,3-dimethoxybenzene. Brominated product 33.11, which demonstrated limited stability upon isolation, was directly oxidized under a continuous-flow process, generating ketone 33.13 via a modified Kornblum oxidation with 2-picoline N-oxide (33.12).

Step 3: Preparation of Belzutifan
Having established 33.13, the remaining steps in the synthesis of belzutifan focused on securing the three contiguous stereocenters present on the indane ring. First, installation of the chiral alcohol in 33.14 was completed by asymmetric reduction under transfer hydrogenation conditions with (R,R)-Ru-diphenylethylenediamine ((R,R)-Ru-DPEN) and HCO2H/TEA in acetonitrile, which provided the desired (R)-hydroxyketone in high selectivity. Immediate quenching of the reduction step with aqueous HCl facilitated cleavage of the ketal moiety, enabling isolation of intermediate 33.14 in 91% overall yield and >99% ee after crystallization from water. The α-fluorination of intermediate 33.14 was performed by reaction with Selectfluor and catalytic methanesulfonic acid, providing a mixture of fluorinated diastereomers (33.15) which were immediately subjected to a Noyori asymmetric transfer hydrogenation process after quenching of excess fluorinating reagent with TEA. Under these hydrogenation conditions (RuCl[(R,R)-TsDPEN](pcymene)/TEA/HCO2H/MsOH), reduction of the ketone occurred with high selectivity while simultaneously facilitating a dynamic kinetic resolution (DKR) process of the neighboring fluoride substituent in a single-pot reaction. While acidmediated ketal side products were observed in this reaction sequence, quenching of the reaction with water enabled isolation of the desired diastereomer 33.16 in 80% yield (99:1 dr) from 33.14. Finally, a deoxyfluorination reaction of intermediate 33.16 with perfluoro-1-butanesulfonyl fluoride (PBSF, 33.17) was used to install the final stereocenter. In this case, the use of DBU in the reaction was essential for selective deprotonation of the desired alcohol, an observation supported by computational studies. This reaction took place with complete inversion of stereochemistry, providing belzutifan in 73% isolated yield (99.8% purity) after recrystallization from water.