Dexmedetomidine Uses, Side Effects, Pharmacokinetics & Metabolism
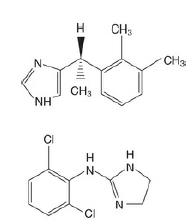
D exmedetomidine is the S -enantiomer of the veterinary anaesthetic medetomidine (Fig.). I t has highly selective activity at α2 adrenoceptors (1600 : 1 α2 : α1) and eight times more affinity than clonidine for this site. D exmedetomidine is licenced in the UK for i.v. sedation of adult patients in intensive care, although there is off-licence use, such as for procedural sedation during an awake craniotomy.
CNS effects
D exmedetomidine has sedative, analgesic and anxiolytic properties and a minimum alveolar concentration (MA C)–sparing effect of up to 90%. Patients given dexmedetomidine require lile or no additional medication to achieve a desired sedative end-point. A unique feature is the ease with which patients can be aroused from an effective level of sedation.
CVS effects
Adverse effects are predominantly cardiovascular (and contributed to by activity at imidazoline receptors as described earlier). Decreases in heart rate, myocardial contractility and systemic vascular resistance reduce myocardial oxygen requirements. This may be advantageous for patients with cardiac risk factors, but undesirable cardiovascular depression may limit use of this agent. Care is required in patients with pre-existing bradycardia or conduction delay because there is a reduction in sympathetic tone and an increase in parasympathetic tone.
Respiratory
There is very little respiratory depression if used as a sole agent.
Endocrine
At therapeutic doses, dexmedetomidine has no significant effect on A CTH secretion, but response to A CTH may be reduced after prolonged use or at high doses.
Metabolism
D exmedetomidine is metabolised via hepatic glucuronidation, and clearance is reduced in patients with liver impairment. Very lile unchanged drug reaches the urine, but 95% of degradation products are excreted this way (4% in faeces). There is a theoretical possibility of accumulation of metabolites in patients with renal failure, but toxicity has not been described because active metabolites of dexmedetomidine have not been identified at present.
Pharmacokinetics
D exmedetomidine is freely soluble in water and has a pKa of 7.1. The pharmaceutical formulation is a clear, colourless preservative-free solution with a pH of 4.5–8. Protein binding of dexmedetomidine is 94%, with negligible displacement by drugs commonly used in anaesthetic and I CU practice. The elimination half-life is approximately 2h and the steady-state volume of distribution is 118 l. There are no significant differences in the pharmacokinetic profile in the elderly, but an enhanced clinical response is seen.
Dosage
Dosage of dexmedetomidine is as follows:
• ICU sedation: infusion at an average of 0.7 μg kg−1 h−1 (normal
range 0.2–1.4 μg kg−1 h−1).
• Procedural sedation: 1 μg kg−1 over 10 min, then infusion at
an average of 0.6 μg kg−1 hr−1 (normal range 0.2–1.0 μg kg−1 hr−1).

![328086-60-8 Methyl (S)-2-(Boc-amino)-3-[(S)-2-oxo-3-pyrrolidinyl]propanoatePropertiesApplicationsRecommended](/NewsImg/2022-02-22/6378112190732321809669208.jpg)