7-Azaindole: A Versatile Scaffold for Developing Kinase Inhibitors
The majority of kinase inhibitors have been developed as ATP competitors to interact with a hinge region in ATP binding sites of kinases. 7-Azaindole has been found as an excellent hinge binding motif by making two hydrogen bonds with the kinase hinge region. Vemurafenib, a B-RAF kinase (serine–threonine kinase [STK]) inhibitor approved by the U.S. Food and Drug Administration (FDA) for the treatment of melanoma, was created from this simple 7-azaindole fragment by successful use of structure-based drug design techniques. The huge potential of 7-azaindole as a hinge-binding motif has encouraged many researchers to employ it as a kinase privileged fragment. This paper will review recent examples of 7-azaindole-based kinase inhibitors, and discusses their binding interactions with the kinase hinge regions.
1. Introduction
7-Azaindole is well known as a kinase privileged fragment,7,8) and has been incorporated into many kinase inhibitors as a hinge-binding element. As shown in Fig. 1a, the pyridine N atom and the pyrrole NH in the 7-azaindole ring serve as a hydrogen bond acceptor and donor, respectively, to make bidentate hydrogen bonds with the hinge region of the kinase. In addition, 7-azaindole has five modification sites where various substituents can be readily attached. Therefore recent years have seen growing interest in this 7-azaindole scaffold; more than 100000 chemical structures having 7-azaindole framework have been registered in the CAS chemical database,9) which is steadily increasing by the synthetic innovation and enrichment of commercially available derivatives.10) Figure 1b shows representative kinase inhibitors having a number of substituents at various positions in the 7-azaindole ring.11–16) Among them, vemurafenib (1), a B-RAF kinase (serine–threonine kinase [STK]) inhibitor, is the first FDA-approved 7-azaindole-based kinase drug for the treatment of melanoma.11) Vemurafenib was discovered through lead optimization starting from a small 7-azaindole fragment, and is recognized as the first successful example of “fragment-based” drug discovery approach.17) Currently, several 7-azaindole-based kinase inhibitors (2–6) are under clinical evaluation as shown in Fig. 1b. Apart from PLX-8394 (6), a second-generation BRAF kinase inhibitor,16) other compounds have been developed targeting various kinds of kinases including Janus kinase 3 (JAK3; a cytoplasmic tyrosine kinase [TK]),12) colony stimulating factor 1 receptor (CSF1R; a receptor TK),13) aurora kinases (STK),14) and rho-associated, coiled-coil-containing protein kinase 1 (ROCK1; STK).15) Comprehensive survey of a drug discovery database (substructure-search using Thomson Reuters IntegritySM database18) as of March 2017) revealed that more than 90 kinds of kinases have been shown sensitive to 7-azaindole-based kinase inhibitors—sufficient to cover the whole human kinome as shown in Fig. 2. These data suggest that 7-azaindole fragment can be useful as a starting point of medicinal chemistry targeting various kinases.
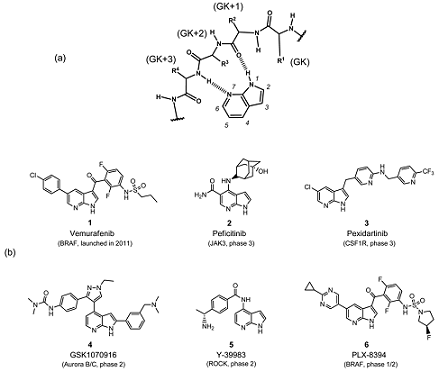
Fig. 1. (a) Hydrogen Bonds between 7-Azaindole Fragment and the Hinge Region of a Kinase Are Indicated by Dashed Line; (b) 7-Azaindole-Based Kinase Inhibitors in Clinic and Clinical Development
GK: gatekeeper amino acid residue.
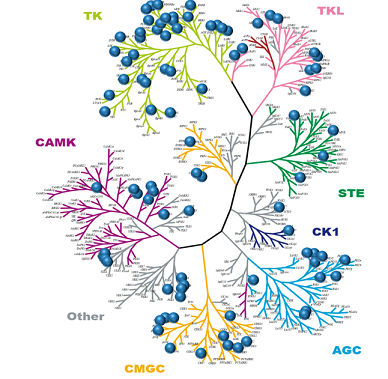
Fig. 2. 7-Azaindole Scaffold Provides Broad Coverage of the Human Kinome
2. Binding Mode of 7-Azaindole-Based Inhibitors
First, we investigated X-ray cocrystal structures of kinases complexed with small molecules having the 7-azaindole fragment available in the RCSB Protein Data Bank (PDB). We found a total of 70 structures registered in the PDB as shown in Table 1. These structural data include 37 kinds of kinases that belong to different kinase families.
Analysis of the captured cocrystal structures revealed that the 7-azaindole group binds to kinases with different binding modes, which can be classified into three groups such as “normal” (azaindole binds to the hinge as most frequently seen), “flipped” (azaindole moiety is flipped 180° with respect to “normal” binding mode), and “non-hinge” (azaindole binds at a different site from the hinge region; Fig. 3). As shown in Fig. 3, “normal” and “flipped” binding modes can form bidentate hydrogen bonds with the backbone amides of hinge amino acid residues, GK+1 (next to the gatekeeper residue) and GK+3 residues. Namely, the carbonyl oxygen of GK+1 interacts as a hydrogen bond acceptor and backbone N–H of GK+3 donates hydrogen bond in “normal” binding mode. On the other hand, in “flipped” binding mode, the GK+3 residue acts as both hydrogen bond donor and acceptor. In the case of inhibitors categorized into “non-hinge” binding mode, the 7-azaindole moiety binds at a different site from the hinge region, because such compounds have another hinge-binding motif in addition to 7-azaindole. The observation that both “normal” and “flipped” binding modes are possible in Janus kinase 2 (JAK2; TK), c-Met (Met; TK), and apoptosis signal-regulating kinase 1 (ASK1; STK) kinases suggests that the binding mode is not dependent on kinase structural features. Rather, it would depend on the ligand structure, because 2-substituted derivatives of the azaindole give only “flipped” binding mode (compounds 7–1119–23) in Fig. 4). As illustrated in Fig. 3, the 2-position of the azaindole ring is directed to the GK residue in “normal” binding mode, and substitution at this position would result in steric hindrance. In contrast, there is open space facing the solvent-exposed space when compounds bind with the “flipped” binding mode, and this would bring a significant preference for binding orientation of 2-substituted azaindoles. Compounds 12–16 also bind to kinases with the “flipped” orientation24–28); these compounds are type II kinase inhibitors, which can interact with a deep allosteric pocket created by conformational change in the activation loop that adopts the inactive-DFG-out conformation. Almost all 7-azaindoles with “normal” binding modes are type I kinase inhibitors except one ligand (PDB code, 3ETA), suggesting that there might be some relation between binding orientation of azaindole and type of kinase inhibitors.
3.Concluding Remarks
After the success of imatinib, protein kinases have become the most important class of drug target, especially in oncology. The majority of kinase inhibitors reported so far bind to the hinge region of kinases in a similar manner, and thus increased understanding the binding mode of the hinge binder moiety will provide new insights into the design of novel kinase inhibitors. Many hinge binding moieties have been reported and incorporated into kinase inhibitors as a hinge binding element. Among them, 7-azaindole has several useful features: possible bidentate hydrogen bond interactions; well-established chemistry for installing substituents into the ring structure; and good commercial availability of starting materials. We conducted a systematic analysis of the binding modes of 7-azaindole-based kinase inhibitors registered in the PDB, and found that those inhibitors are classified into three binding modes, such as “normal,” “flipped,” and “non-hinge,” based on the crystal structures complexed with the target kinases. It is noteworthy that these binding modes could be easily switched among each other by small modifications of the inhibitors, and this phenomenon could confuse medicinal chemists. Therefore it is important to confirm the binding mode by X-ray structural analysis throughout SAR studies.
You may like
Related articles And Qustion


See also


Lastest Price from 7-Azaindole manufacturers

US $9.85/KG2025-04-21
- CAS:
- 271-63-6
- Min. Order:
- 1KG
- Purity:
- 99%
- Supply Ability:
- 10 m
US $0.00-0.00/KG2025-04-04
- CAS:
- 271-63-6
- Min. Order:
- 1KG
- Purity:
- 98%
- Supply Ability:
- 1Ton