

一、研究背景
增殖性视网膜病变会引发视网膜初始血管缺失与神经组织缺血,机体为恢复代谢稳态会大量分泌血管内皮生长因子,最终诱发病理性新生血管生成,严重威胁患者视力。目前针对该疾病的治疗手段主要以抑制血管内皮生长因子为主,仅能遏制病理性血管生长,无法实现缺血视网膜的生理性再血管化。 过往研究已筛选出多种调控病理性血管生成的分泌因子,但血管微环境的代谢变化能否决定新生血管向病理或生理方向分化,仍缺乏明确论证。厘清病理性新生血管与生理性新生血管的分子及代谢特征,解析血管微环境对血管生成命运的调控机制,是实现视网膜损伤修复、保护神经元功能的关键。
二、研究材料与方法
该研究同时纳入增殖性糖尿病视网膜病变患者临床样本与氧诱导视网膜病变小鼠模型,综合运用代谢物检测、转录组测序、单细胞测序、基因敲除、药物干预以及视网膜功能检测(ERG的PhNR)等多项实验技术,系统分析病变组织的代谢特征、基因表达模式以及血管形态与视网膜功能变化。
三、研究结果
3.1 增殖性视网膜病变以脂肪酸氧化为核心代谢特征
研究人员对患者玻璃体样本与模型小鼠视网膜组织开展代谢物及转录谱分析,结果显示,两类样本中酰基肉碱等脂肪酸氧化代谢物均出现显著累积,脂肪酸代谢相关通路在病变组织中占据主导地位。单细胞测序结果进一步证实,视网膜血管单位相关细胞,包括内皮细胞、星形胶质细胞、周细胞等,均高表达Acadl、Hadha等脂肪酸氧化相关基因,明确脂肪酸氧化是增殖性视网膜病变病理性血管生成的标志性代谢通路。
3.2 抑制脂肪酸氧化存在局限性,Sirt3 是调控代谢通路的关键靶点
研究人员使用etomoxir抑制脂肪酸氧化关键酶CPT1a,虽可减少病理性新生血管丛,但该药物会引发明显全身毒性,导致缺氧阶段小鼠死亡率上升,无法应用于临床。线粒体脂肪酸氧化调节因子Sirt3在病变星形胶质细胞中表达水平显著上调,成为后续研究的核心靶点。 实验发现,敲除Sirt3基因后,小鼠视网膜病理性血管丛数量、血管闭塞面积均明显下降,且未产生明显毒副作用。同时,视网膜整体代谢模式发生转变,脂肪酸氧化被抑制,糖酵解通路被激活,组织葡萄糖摄取量增加,星形胶质细胞内血管内皮生长因子表达水平同步升高。这表明Sirt3缺失能够推动新生血管微环境从脂肪酸氧化向糖酵解转变。
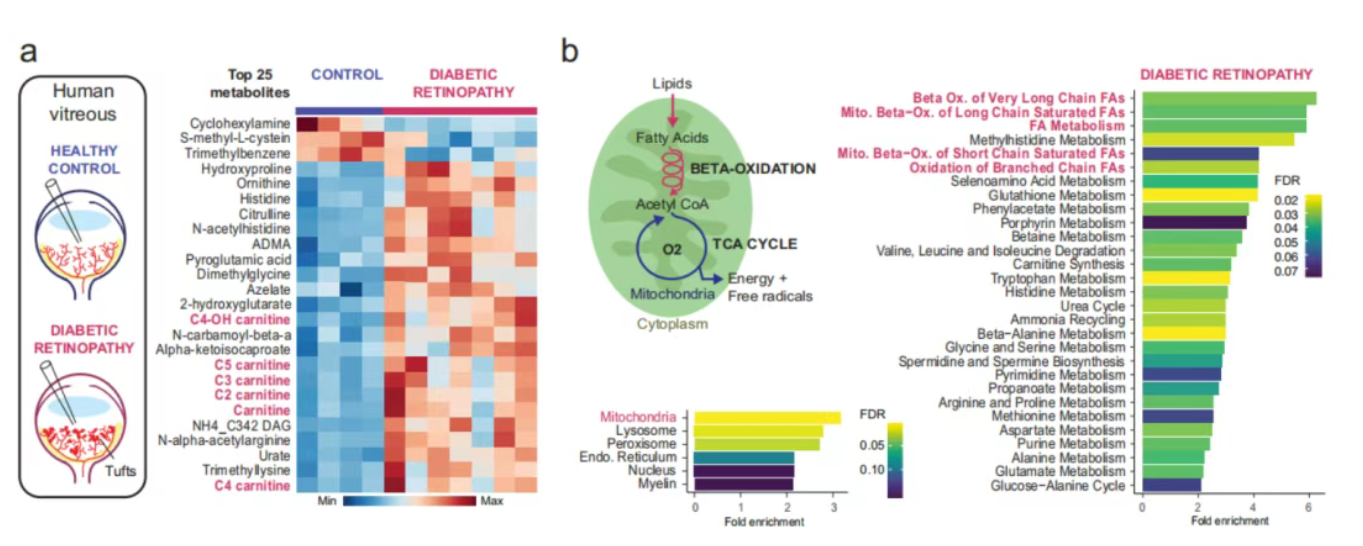
脂肪酸氧化是人类增殖性糖尿病视网膜病变的显著特征之一
3.3 Sirt3 缺失重编程血管表型,促进视网膜再生与功能修复
单细胞转录组分析显示,Sirt3敲除后,早期病理性血管丛的内皮细胞转录特征逐步向生理性尖端细胞转变,尖端细胞标志物与糖酵解相关基因表达上调,细胞丝状伪足数量增多,有效加快了缺血视网膜的再血管化进程。 视网膜功能检测结果印证,Sirt3缺失小鼠的神经视网膜功能得到显著改善。综上,靶向敲除Sirt3可将病理性血管丛重编程为具备再生能力的生理性血管表型,最终实现增殖性视网膜病变的治疗效果。
四、讨论与结论
该研究完整阐明了增殖性视网膜病变中病理性新生血管的代谢机制:病理性新生血管的生存与增殖高度依赖脂肪酸氧化代谢。敲除代谢调节因子Sirt3可完成新生血管微环境的代谢重编程,将主导代谢通路由脂肪酸氧化切换为糖酵解,一方面抑制病理性血管丛形成,另一方面诱导产生生理性再生血管,修复缺血损伤的视网膜,并改善视网膜神经功能。 该研究首次区分了病理性新生血管与生理性再生血管的代谢差异,打破了传统单纯抑制血管生成的治疗思路,提出通过调控血管微环境代谢、诱导血管表型转换来治疗血管类疾病的全新策略。Sirt3可作为增殖性视网膜病变极具潜力的治疗靶点,也为其他缺血性血管疾病的机制研究与临床干预提供了重要的理论支撑与实验依据。
五、血管微环境研究检测服务哪个公司有?
LabEx 可提供专业的视网膜血管疾病研究相关检测技术服务。平台拥有Luminex/MSD多因子检测、代谢组检测、转录组测序、单细胞测序等多项成熟技术平台,可完成视网膜病变代谢特征分析、血管内皮细胞转录谱解析、疾病标志物筛选、通路机制验证及体内功能评价等实验项目,全面适配视网膜血管再生机制探究、代谢重编程机制解析、疾病靶点验证与药效评估等各类科研需求。
