| Anti-cancer drugs |
On May 20, 2014, Clovis Oncology announced that the US FDA had granted its test drug CO-1686 for breakthrough treatment drug qualification, as a second line, single administrated drug for the treatment of EFGR mutation non-small cell lung cancer (NSCLC) of the T790M mutation patients. The awarding for this breakthrough therapeutic drug eligibility was based on the efficacy and safety results of Co-1686 in an ongoing Phase 1/2 study. Data of related study have shown that CO-1686 is a third-generation EGFR inhibitor, being used for the treatment of non-small cell lung cancer, overcoming the drug resistance generated from the EGFR T790M mutations, and has excellent efficacy and tolerability on NSCLC with T790M + EGFR mutations. Its major market competitor, the third-generation EGFR inhibitor, AZD9291 (AstraZeneca) has also gained FDA's groundbreaking drug eligibility.
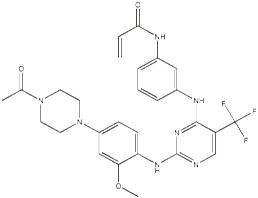
CO-1686 is a novel, oral-administrated, targeted covalent (irreversible) inhibitor of the epidermal growth factor receptor (EGFR) mutations, being able to suppress key activation mutations and T790 drug-resistant mutations, leaving the wild-type EGFR signal unused. This drug was developed for the treatment of NSCLC patients carrying initially activated EGFR mutations and major resistant mutant T790M.
The above information is compiled and edited by Xiao Nan of Chemicalbook. |
| Biological activity |
Rociletinib (CO-1686, AVL-301) is an irreversible, mutation-selective EGFR inhibitor that targets EGFRL858R/T790M and EGFRWT with a Ki of 21.5 nM and 303.3 nM, respectively. Phase 2.
Target: EGFR (L858R/T790M) EGFR (wt)
IC50: 21.5 nM (Ki) 303.3 nM (Ki) |
| In vitro study |
CO-1686 inhibited the p-EGFR in EGFR-expressing cells with an IC50 ranging from 62 to 187 nM while inhibiting EGFR phosphorylation. In three WT EGFR-expressing cells, the IC50 is larger than 2,000 nM. CO-1686 can selectively inhibit the growth of mutant EGFR expressing NSCLC cells with a GI50 ranging from 7 to 32 nM while inducing apoptosis. The CO-1686-resistant NSCLC cell line exhibited a signal for epithelial-mesenchymal transition and increased susceptibility to AKT inhibitors. |
| In vivo studies |
In all EGFR mutant models, as well as in transgenic mice expressing human EGFRL858R-and EGFRL858R/T790M, CO-1686 caused significant tumor growth inhibition in a dose-dependent manner. |
| Synthesis method |
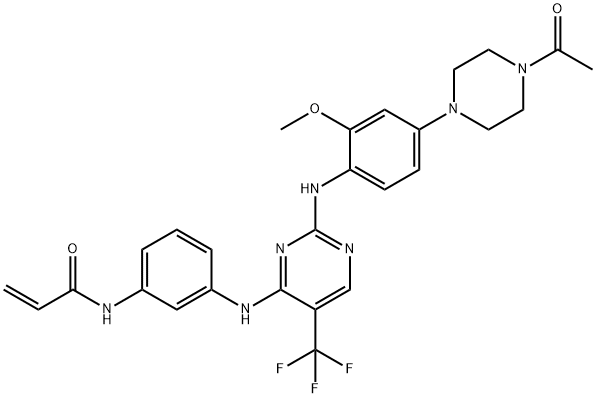
5-fluoro-2-nitroanisole was condensed with piperazine, acetylated and reduced to give 1-(3-methoxy-4-aminophenyl)-4-acetylpiperazine (4) In addition, use 2, 4-dichloro-5-trifluoro methyl pyrimidine to undergo condensation with 3-nitroaniline, reduction and amidation to obtain N-[3-(2-Chloro-5-trifluoromethyl-pyrimidin-4-amino) phenyl] acrylamide (7). 4 and 7 were condensed to give the EGFR inhibitor, the anticancer drug CO-1686 with a total yield of about 71% (based on 2, 4-dichloro-5-trifluoromethyl pyrimidine).
Reference: Synthesis of CO-1686 [J]. Chinese Pharmaceutical Industry, 2014, 45 (8): 710-713.
OF: (1) Lai Yisheng, male, professor, doctoral supervisor, engaged in anti-inflammatory and anti-tumor drugs studied. (2) Zhang Shan, female, graduate students, professional direction: medicinal chemistry. |

 China
China