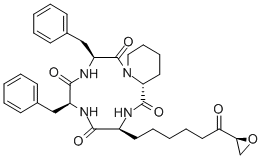
Trapoxin A is a cyclotetrapeptide histone deacetylase (HDAC) inhibitor. It inhibits HDAC activity in a concentration-dependent and irreversible manner. Trapoxin A inhibits proliferation of NIH3T3 cells transformed by the oncogene v-sis (sis/NIH3T3; IC50 = ~200 ng/ml) and has detransformation activity, flattening sis/NIH3T3 cells when used at a concentration of 1 ng/ml. It also halts the cell cycle at the G2 phase in 3Y1 fibroblasts when used at a concentration of 10 nM and inhibits growth of TR303 cells resistant to trichostatin A at a concentration of 50 ng/ml.
Trapoxin A has been used:
- to study its effects on the inhibition of histone deacetylase 11 (HDAC11)
- to study its effects on the inhibition of HDAC3 in human cell lines
- to study its effects on the inhibition of HDAC6 in rat pyramidal neurons
ChEBI: A homodetic cyclic tetrapeptide constructed from L-phenylalanyl (x2), D-pipecolinyl and L-2-amino-8-oxo-9,10-epoxydecanoyl residues.
Trapoxin A is a cyclotetrapeptide and a histone deacetylase (HDAC) inhibitor. It increases the level of chromatin acetylation associated with histone H3 at low nanomolar concentrations. Unlike the reversible HDAC inhibition induced by TCA, Trapoxin A irreversibly inhibites HDAC activity in crude cell lysates, and induces the accumulation of hyperacetylated core histones in a number of mammalian cell lines and tissues. Histone acetylation and methylation have been studied extensively for their anti-tumor activities in carcinogenesis and Trapoxin has been suggested as a potential anticancer agent for pre-clinical trials.