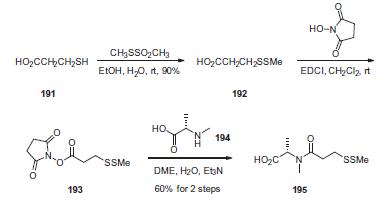
First, commercial 3-mercaptopropionic acid (191) was treated
with methanethiolsufonate to give the corresponding methyldithio
analog 192 in 90% yield. Activation of the acid with N-hydroxysuccinimide
in the presence of 1-ethyl-3-(3-dimethylaminopropyl)-
carbodiimide (EDCI) provided the activated ester 193, which was
reacted with N-methyl-L-alanine (194) to give acid 195 in 60% yield
from compound 192.
Preparation of the DM1 linker-payload is described in
above. The starting material used for the production of DM1
is ansamitocin P-3 (196), which is produced via fermentation
of the microorganism Actinosynnema pretiosum. The ester group
of 196 was removed using a reductive process in the presence of
lithium trimethoxyaluminum hydride to give maytansinol 197 in
85% yield. The use of reductive conditions was required to
avoid subsequent elimination to the a,b-unsaturated amide.
Esterification with 195 in the presence of 1,3-dicyclohexylcarbodiimide
(DCC) and zinc chloride provided DM1¨CSMe 198 in 32%
yield. Reductive removal of the dithiane using dithiothreitol
(DTT) in aqueous buffer at pH 7.5 gave DM1 thiol 199 in 76% yield,
which was utilized in the conjugation to trastuzumab (200).

Completion of the synthesis of trastuzumab emtansine is
described in above. The surface accessible lysine residues of trastuzumab (200) were treated with succinimidyl-4-(N-maleimidomethyl)-
cyclohexane-carboxylate (SMCC, 201) in pH 7.0 buffer
to give amide 202 with approximately four SMCC molecules added
per antibody in 88% yield. Next, the free thiol group of
DM1 (199) was conjugated to the maleimide groups present on
202 to give trastuzumab emtansine (XXV) with an average 3.5
drug molecules loaded per antibody.

