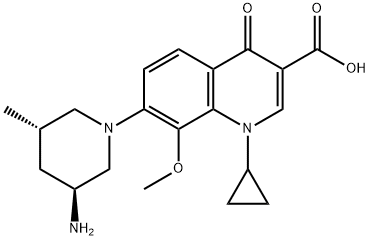
Although several synthetic approaches to marketed quinolone
antibiotics similar in structure to nemonoxacin have been
reported, two dedicated synthetic routes to nemonoxacin
have been reported. Condensation of commercial 2,4-
difluoroacetophenone (57) with ethylene glycol furnished ketal
58 in 86% yield. This was followed by fluorine-directed olithiation
with n-butyllithium and trimethylborate quench.
Acidification followed by oxidation of the boron species
rendered hydroxyketone 59 in 79% yield from 58. Next,
phenol methylation with dimethyl sulfate followed by
deprotonation and reaction with diethyl carbonate (60) gave
rise to the keto-ester intermediate, which underwent condensation
with dimethylformamide-dimethylacetal (DMFDMA)
in refluxing toluene to provide the corresponding
vinylogous amide 61. An addition-elimination reaction with
cyclopropylamine (55) and subjection of this intermediate to
acetimidate 62 in refluxing toluene presumably facilitated
alkene isomerization with concomitant cyclization to produce
the quinolinone derivative 63 in 82% yield over five steps.
Acidic hydrolysis followed by treatment with diboron trioxide
and acetic anhydride generated triacetoxyborate 64, which
served as a unique protecting group for the next step of the synthesis. Exposure of 64 to aminopiperidine 65 under SNAr conditions
provided aniline derivative 66. This was followed by basemediated
borate removal, acidic quench with concomitant Boc
deprotection, and basification to furnish nemonoxacin (V) in
79% yield from 64.
For the preparation of aminopiperidine fragment 65 of
nemonoxacin, commercial proline derivative 67 was converted
to the corresponding ester 68 in 52% yield prior to treatment
with Bredereck?ˉs reagent to give enamine 69 .
Next, catalytic hydrogenation of 69 using a Pfaudler reactor and
5% Pd/C converted the vinylogous amide to the corresponding
methyl group, delivering 70 in nearly quantitative yield and
93:7 diastereomeric excess in favor of the desired geometry.
Further reduction of 70 using NaBH4 followed by treatment
with calcium chloride dihydrate gave the corresponding diol 71 in 66% yield. Mesylation of diol 71 followed by cyclization with
benzylamine and hydrogenation to remove the N-benzyl group
provided aminopiperidine 65. The yields of the last three
steps were not reported.