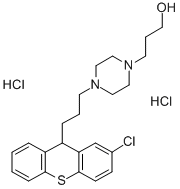
Usage And Synthesis
A stirred mixture of 50.0 g (0.32 mole) of commercial o-mercaptobenzoic
acid, 200 ml of chlorobenzene and 500 ml of 96 % sulfuric acid was stirred at
approximately 70°C for 6 h. After the mixture had remained overnight at
25°C, the layers were separated.
Ice cubes were added to the sulfuric acid layer and the water formed was allowed to diffuse slowly into the sulfuric acid. The finely divided solid was collected on a filter, washed with water by trituration, and refiltered. The filter cake was digested in 300 ml of hot isopropyl alcohol, filtered, and the 3: 10- (2-chloro)thiaxanthenone, melting point 151-152°C (recrystallized from hot dimethylformamide) was obtained. Yield, 32.0 g (41 %).
As a structure proof, the compound was prepared by an alternate route: 5- chloroanthranilic acid, prepared by the chlorination of anthranilic acid, according to J. Am. Chem.Soc. 68, 1303 (1946) was converted into 5-chloro- 2-mercaptobenzoic acid by the method described in J. Org. Chem., 18, 1380 (1953), and the latter was condensed with benzene by means of sulfuric acid to yield 10-(2-chloro)thiaxanthenone, identical with that obtained by the first method.
A stirred mixture of 20.0 g (0.081 mole) of 10-(2-chloro) thiaxanthenone, 20.0 g of red phosphorus and 125 ml of acetic anhydride was treated dropwise with 125 ml of hydroiodic acid (D=1.5). The reaction was extremely exothermic, and after the addition, the mixture was stirred and refluxed for 6 h. It was then cooled somewhat and poured into ice-water. The mixture was filtered and the filter cake was treated with boiling methanol and filteredhot. The pale yellow filtrate was concentrated, cooled, and the 4: 2- chlorothiaxanthene, melting point 102-103°C (recrystallization from ethanol) was isolated. Yield, 11.0 g (59 %).
This compound was also prepared by alternate procedure: 10-(2- chloro)thiaxanthenone was reduced to10-(2-chloro)thiaxanthenol with sodium amalgam in methanol according to the procedure described for 10- thiaxanthenone to 10-thiaxanthenol in J. Am. Chem. Soc., 72, 5332 (1950). The 10-(2-chloro)thiaxanthenol, so obtained, melted at 128-129 °C centigrade and was disproportionated into a mixture of 10-(2-chloro)thiaxanthenone and 2-chlorothiaxanthene by reflexing in glacial acetic acid for 2 h. The mixture was separated by fractional crystallization from methanol in which the 2- chlorothiaxanthene is the more soluble.
To a solution of butyl lithium, prepared from 1.2 g (0.17 g) of lithium, 10 ml of butyl bromide and 200 ml of dry ether according to J. Am. Chem.Soc. 71, 1499 (1949), washdded 3.1 g (0.035 mole) of 2-chlorothiaxanthene and the mixture was stirred under reflux for 3 h under an atmosphere of dry nitrogen. The dark red mixture was then siphoned, in portions, by means of nitrogen pressure into a stirred solution of 80.0 g (0.4 mole) of 1,3-dibromopropane in 300 ml of dry ether. During the addition, which was over a period of 15 min, the deep red color of the organo-metallic solution was immediately discharged and a white precipitate formed. The mixture was stirred and refiuxed for 1 h, filtered, and the filtrate was washed with water, then with dilute hydrochloric acid and dried over magnesium sulfate. Fractional distillation yielded, in addition to the unreacted 1,3-dibromopropane, 4.7 g (52%) of 2-chloro-10-(3-bromopropyl)thiaxanthene as a viscous oil, boiling point 190-204°C (0.7 millimeters).
A mixture of 7.6 g of 2-chloro-10-(3-bromopropyl)thiaxanthene, 5.8 g of 1-(3- hydroxypropyl)piperazine, 6.0 g of anhydrous potassium carbonate and 50 ml of toluene was refluxed for 16 h. Water was added to the cooled mixture and the layers were separated. The toluene solution was extracted with dilute hydrochloric acid, the acidic aqueous solution was made alkaline with sodium hydroxide, and the oil was extracted with ether. The ether solution was washed with water, dried and the 2-chloro-10-{3-[1-(3-hydroxypropyl)-4- piperazinyl]propyl}thiaxanthene was obtained.
Ice cubes were added to the sulfuric acid layer and the water formed was allowed to diffuse slowly into the sulfuric acid. The finely divided solid was collected on a filter, washed with water by trituration, and refiltered. The filter cake was digested in 300 ml of hot isopropyl alcohol, filtered, and the 3: 10- (2-chloro)thiaxanthenone, melting point 151-152°C (recrystallized from hot dimethylformamide) was obtained. Yield, 32.0 g (41 %).
As a structure proof, the compound was prepared by an alternate route: 5- chloroanthranilic acid, prepared by the chlorination of anthranilic acid, according to J. Am. Chem.Soc. 68, 1303 (1946) was converted into 5-chloro- 2-mercaptobenzoic acid by the method described in J. Org. Chem., 18, 1380 (1953), and the latter was condensed with benzene by means of sulfuric acid to yield 10-(2-chloro)thiaxanthenone, identical with that obtained by the first method.
A stirred mixture of 20.0 g (0.081 mole) of 10-(2-chloro) thiaxanthenone, 20.0 g of red phosphorus and 125 ml of acetic anhydride was treated dropwise with 125 ml of hydroiodic acid (D=1.5). The reaction was extremely exothermic, and after the addition, the mixture was stirred and refluxed for 6 h. It was then cooled somewhat and poured into ice-water. The mixture was filtered and the filter cake was treated with boiling methanol and filteredhot. The pale yellow filtrate was concentrated, cooled, and the 4: 2- chlorothiaxanthene, melting point 102-103°C (recrystallization from ethanol) was isolated. Yield, 11.0 g (59 %).
This compound was also prepared by alternate procedure: 10-(2- chloro)thiaxanthenone was reduced to10-(2-chloro)thiaxanthenol with sodium amalgam in methanol according to the procedure described for 10- thiaxanthenone to 10-thiaxanthenol in J. Am. Chem. Soc., 72, 5332 (1950). The 10-(2-chloro)thiaxanthenol, so obtained, melted at 128-129 °C centigrade and was disproportionated into a mixture of 10-(2-chloro)thiaxanthenone and 2-chlorothiaxanthene by reflexing in glacial acetic acid for 2 h. The mixture was separated by fractional crystallization from methanol in which the 2- chlorothiaxanthene is the more soluble.
To a solution of butyl lithium, prepared from 1.2 g (0.17 g) of lithium, 10 ml of butyl bromide and 200 ml of dry ether according to J. Am. Chem.Soc. 71, 1499 (1949), washdded 3.1 g (0.035 mole) of 2-chlorothiaxanthene and the mixture was stirred under reflux for 3 h under an atmosphere of dry nitrogen. The dark red mixture was then siphoned, in portions, by means of nitrogen pressure into a stirred solution of 80.0 g (0.4 mole) of 1,3-dibromopropane in 300 ml of dry ether. During the addition, which was over a period of 15 min, the deep red color of the organo-metallic solution was immediately discharged and a white precipitate formed. The mixture was stirred and refiuxed for 1 h, filtered, and the filtrate was washed with water, then with dilute hydrochloric acid and dried over magnesium sulfate. Fractional distillation yielded, in addition to the unreacted 1,3-dibromopropane, 4.7 g (52%) of 2-chloro-10-(3-bromopropyl)thiaxanthene as a viscous oil, boiling point 190-204°C (0.7 millimeters).
A mixture of 7.6 g of 2-chloro-10-(3-bromopropyl)thiaxanthene, 5.8 g of 1-(3- hydroxypropyl)piperazine, 6.0 g of anhydrous potassium carbonate and 50 ml of toluene was refluxed for 16 h. Water was added to the cooled mixture and the layers were separated. The toluene solution was extracted with dilute hydrochloric acid, the acidic aqueous solution was made alkaline with sodium hydroxide, and the oil was extracted with ether. The ether solution was washed with water, dried and the 2-chloro-10-{3-[1-(3-hydroxypropyl)-4- piperazinyl]propyl}thiaxanthene was obtained.
Preparation Products And Raw materials
PROMPT×
PROMPT
The What'sApp is temporarily not supported in mainland China
The What'sApp is temporarily not supported in mainland China
Cancel
Determine