5-(戊烷-3-基氧基)-7-氧代-双环[4.1.0]庚-3-烯-3-羧酸乙酯的制备
发布日期:2020/9/25 9:55:06
背景及概述[1]
5-(戊烷-3-基氧基)-7-氧代-双环[4.1.0]庚-3-烯-3-羧酸乙酯是磷酸奥司米韦(oseltamivirphosphate,1)的中间体。磷酸奥司米韦是美国GileadSciences公司研发的神经氨酸酶抑制剂,1999年在美国和瑞士等国首次上市,商品名Tamiflu(达菲)。本品为口服制剂,主要通过干扰病毒从被感染宿主细胞表面的释放减少病毒传播,临床用于预防和治疗A、B型流感病毒导致的流行性感冒,是预防和治疗H5N1型禽流感的首选药物。
制备[2]
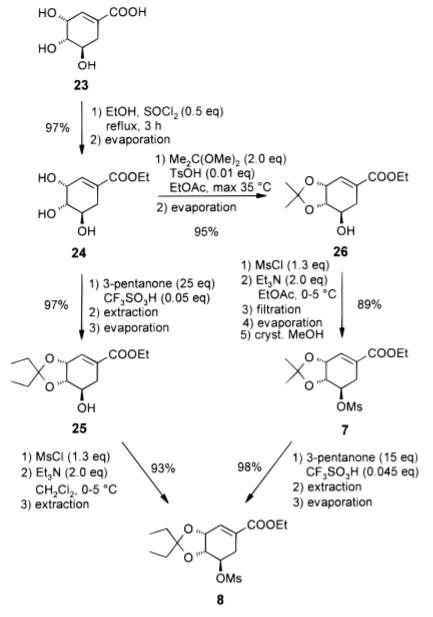
步骤一、(3R,4S,5R)-3,4,5-三羟基-环己-1-烯-羧酸乙酯(24)的合成
在30分钟内向莽草酸(2.00kg,10.5mol)的EtOH(8.0L)中加入SOCl2(0.61kg,5.13mol),然后加入甲苯(0.8L)冲洗管路。在添加SOCl2的过程中温度从室温升至35°C。完成后另外,将反应混合物加热至沸腾温度3小时。约1小时后,所有莽草酸溶解,得到深棕色溶液。在45分钟内将反应混合物冷却至40°C,在此温度(100mbar)下减压浓缩。反应混合物的GC分析表明剩余1.7%的莽草酸,产物24有90.7%。24为粘性棕色油(2.2-2.7升)的形式,含有约20%的乙醇,用于下一步无需进一步纯化。
步骤二、3,4-异亚丙基莽草酸乙酯(26)的合成
向粗制的莽草酸乙酯24中,加入EtOAc(13.6L),2,2-二甲氧基丙烷(1.070kg,10.3mol)和TsOH·H2O(0.020kg,0.105mol)。在150-200mbar的真空下,反应混合物用50°C夹套加热至沸腾温度(30-35°C)。蒸出溶剂,约2小时后,初始体积减半。加入第二当量的2,2-二甲氧基丙烷(1.070kg,10.3mol),并在30-35°C下继续进行蒸馏2小时。然后将反应混合物在20mbar下蒸发至干获得26,为深棕色油状物。无需进一步纯化即可用于下一步。
步骤三、3,4-O-异亚丙基-5-O-(甲磺酰基)莽草酸乙酯(7)的制备
向26在EtOAc(15.2L)中的溶液中,加入MsCl(1.52kg,13.3mol)。然后,加入Et3N(2.32kg,22.9mol),内温不得超过20°C。将混合物在20°C下再持续搅拌30分钟。反应混合物的GC分析显示0.13%的26和85.7%的7。将在添加Et3N时形成的沉淀离心去除(Et3N盐酸盐和不溶物),并用EtOAc(3×2.5L)洗涤。合并的有机溶液在低于30°C和100mbr下蒸发至干。将部分结晶的残留物悬浮在MeOH(3.6L)中,并将悬浮液搅拌5分钟。再次通过在100mbr下蒸馏除去溶剂,并将残余物在45-50℃下溶于MeOH(12.0L)中。将溶液以0.33°C/分钟的速度冷却至-20°C。在30-35°C的温度下诱导析晶(10g)。将悬浮液在-20°C下搅拌2小时。离心收集产物并用MeOH(3.6L,-20℃)洗涤。它被干燥到恒重(35°C,20mbar,6h)得到2.78kg甲磺酸盐7(HPLC测定99.7%w/w,8.65mol,82%),米色固体。
步骤四、3,4-O-异亚戊基-5-O-(甲磺酰基)莽草酸乙酯(8)的制备
在100-150mbar的部分真空下,将7(7.50kg,HPLC测定97.7%w/w,22.9mol)和CF3SO3H(0.153kg,1.02mol)的3-戊酮(12.0L)溶液加热至沸腾温度(40°C)。1小时内通过蒸馏除去丙酮和3-戊酮的混合物(1.5L)。然后将真空度调整为50mbar,继续蒸馏4小时。在这时间,以4.5L/h的速率连续添加3-戊酮使反应器中的体积保持恒定。在15分钟内将反应混合物冷却至室温。GC分析显示98.2%的8和0.2%的7。反应混合物用Et3N(0.216kg;2.13mol)中和并在低于35°C,减压下(20mbar)浓缩下。剩余的粗产物中的3-戊酮(约3%)通过与环己烷(2×9.0L)在150-50mbar和30°C下共沸去除。剩余的油溶解在CH2Cl2(6.0L)中,溶液用碳酸氢钠水溶液(7.5%,13.5公斤)萃取。用H2O(8.1L)洗涤有机层,减压浓缩(200-50mbar,30°C),得到亚戊基酮8,黄色油,产率为98%(7.97千克,HPLC分析为98.7%w/w,22.6mol)。
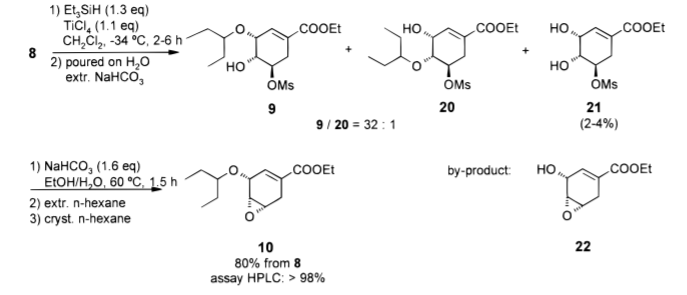
步骤五、乙基(3R,4R,5R)-3-(1-乙基丙氧基)-4-羟基-5-甲磺酰氧基-环己-1-烯-1-羧酸酯(9)的制备
向冷却至-32至-36°C的亚戊基缩酮8(2.63kg,7.55mol)的CH2Cl2(21.0L,H2O含量0.02%)中加入Et3SiH(1.18kg,10.2mol)。以使反应混合物的温度不超过-32℃的速率加入TiCl4(1.60kg,8.47mol)的CH2Cl2(1.95L)溶液。90分钟后完成TiCl4的添加。然后在-36至-32℃下继续搅拌另外2小时。为了监测反应进程,如下所述提取30mL反应混合物样品并淬灭。有机层的HPLC分析表明所有起始原料8已被消耗(0.20%)。将反应混合物快速倒入搅拌的冰/H2O混合物(9.4kg,17.0L)中,并将连接线用CH2Cl2(2.5L)洗涤。所得两相混合物的温度为-3℃,并在35℃的夹套温度下加热至5℃。有机层用NaHCO3水溶液(7.5%,18.4kg)洗涤,并在减压(400-200mbar,35°C)下浓缩。
步骤六、5-(戊烷-3-基氧基)-7-氧代-双环[4.1.0]庚-3-烯-3-羧酸乙酯(10)的制备
向搅拌的粗羟基醚9在EtOH中的溶液中加入NaHCO3水溶液(7.5%,14.0kg)。在60分钟内将反应混合物加热至60-65℃,并在该温度下继续搅拌持续1.5小时。此时间后,HPLC显示所有起始原料均已消耗完(9<0.10%)。在30分钟内将反应混合物冷却至35℃,并用正己烷(4×17.3L)萃取。将合并的有机层在35°C下用H2O(10.2L)洗涤一次,并在减压(200mbar,35°C)下浓缩至残留体积28L。将浅黄色溶液在3小时内冷却至-18°C。在15°C,产品自发结晶。将悬浮液在-18℃下搅拌2h。离心收集产物,并用冷(-15℃)正己烷(2.2L)洗涤。将其干燥(30℃,20mbar,4h)至恒重,得到1.55kg10(蓬松的白色固体)(6.03mol,HPLC测定98.9%w/w,产率80%)。
参考文献
[1]李行舟,谢云德,钟武,etal.磷酸奥司米韦的合成[J].中国医药工业杂志,2007,38(6):393-397.
[2]Org.Proc.Res.Dev.1999,3,4,266–274


欢迎您浏览更多关于5-(戊烷-3-基氧基)-7-氧代-双环[4.1.0]庚-3-烯-3-羧酸乙酯的相关新闻资讯信息