普卢利沙星的制备方法
发布日期:2022/2/8 14:13:48
背景及概述[1]
喹诺酮类药是二十世纪八十年代迅速发展起来的抗菌药,是抗感染药物中开发较活跃的一类药物。喹诺酮类药为高效杀菌药,其杀菌机理是以细菌的DNA为作用靶,通过阻碍DNA拓扑异构酶使细菌DNA无法形成超螺旋,进一步造成染色体的不可逆损害,导致细菌细胞无法分裂繁殖。普卢利沙星是由日本新药公司和明治制果公司共同研制开发研究的新一代氟喹诺酮类抗菌药,于2002年7月获准在日本上市。
普卢利沙星是日本新药公司和明治制果公司共同研发的第四代喹诺酮类抗感染药,该药于2002年7月在日本首次批准,剂型为片剂,规格为132mg(相当于普卢利沙星活性成分100mg),临床用于治疗革兰氏阳性菌及阴性菌引起的呼吸道感染、泌尿生殖系统感染、耳鼻科感染、胆道感染、感染性肠炎、细菌性痢疾、皮肤软组织感染及外科感染等。

制备[1-3]
报道一、
(1)式(III)化合物的制备
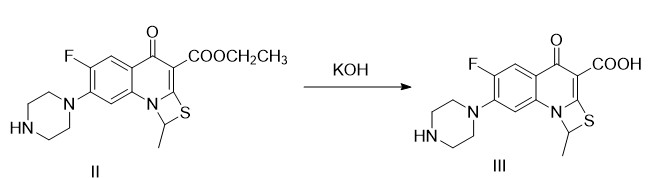
在反应釜中加入8.0kg式(II)化合物,104kg水,5.6kg氢氧化钾,加热至60~70℃进行水解反应,2~3小时。反应完毕后,冷却至室温,28.8kg乙酸乙酯洗涤,分出水层,搅拌下,以浓盐酸调至pH=6~7,继续搅拌0.5小时,抽滤得滤饼,适量乙酸乙酯洗涤滤饼,甩干,收集滤饼,60~70℃热风循环烘干,即得式(III)化合物成品7.32kg,收率(摩尔)94.8%,纯度96.2%;
(2)式(V)化合物的制备
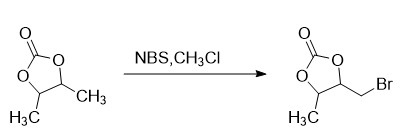
在反应釜中加入5.0kg式(IV)化合物,7.5kgN-溴代琥珀酰亚胺(NBS),0.25kg偶氮二异丁腈,加入90L氯仿为反应溶剂,搅拌升温至38℃,待反应体系稳定,再缓缓升温至回流反应,2-3小时,反应完毕后,冷却至室温,过滤除去不溶物,滤液先常压回收氯仿,4℃冷藏析晶过夜后再次过滤除去不溶物,然后减压蒸馏收集110℃~120℃/5mmHg的馏分,得式(V)化合物8.56kg,收率(摩尔)91.4%,纯度93.8%;
(3)式(I)化合物的制备
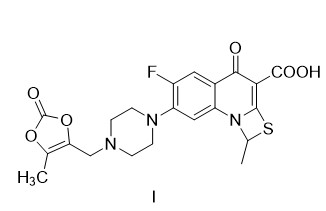
于反应釜中加入7.0kg式(III)化合物、2.1kg碳酸氢钾和42LN,N-二甲基甲酰胺,冷却降温至0℃,滴加浓度为0.6kg/L式(V)化合物的DMF溶液11.7L,控制内温0℃,滴毕,0℃搅拌,反应时间为5小时,将反应液搅拌下倾入到冰水中,搅拌0.5小时,滤集结晶,水洗滤饼至中性,抽干,60~70℃热风循环烘干,得式(I)化合物普卢利沙星的粗品9.42kg,收率(摩尔)95.4%,纯度90.5%;
(4)在反应釜中加入步骤(3)中得到的普卢利沙星的粗品和358L乙腈,搅拌升温回流溶解至透明,稍冷,加入0.05kg活性炭,保温搅拌回流30分钟,趁热过滤,滤液自然冷却至室温结晶,再通冷冻水冷却析晶过夜,离心,滤饼用少量冷冻乙腈洗晶,甩干,80℃真空干燥至干,得普卢利沙星成品8.73kg,收率(摩尔)92.4%,纯度99.5%。
报道二、
DMDO-Cl(2.24g,15.1mmol)溶解在5.5mlDMF中,加入碘化钠(3.28g,22.0mmol),303K搅拌1小时,加入11ml丙酮,继续搅拌1小时,反应液不经分离,往反应液中加入32mlDMF、化合物Ⅱ(5.00g,11.6mmol)、碳酸氢钾(2.69g,26.9mmol),293K搅拌3小时,之后反应液倒入120ml冰水中,析出沉淀,过滤,真空干燥得4.93g普卢利沙星粗品,收率为92.1%,纯度为94.2%。
报道三、
步骤1:
在三口瓶中加入镁条、无水THF与碘(引发剂),然后在加热回流下,慢慢滴加式(II)化合物4-溴甲基-5-甲基-1,3-二氧杂环戊烯-2-酮(作为起始原料)的THF溶液,回流搅拌。快速加入式(III)的乙醛,继续回流反应。加热浓缩,残余物冷却搅拌下缓慢加入冰水和乙酸乙酯,抽滤,滤液分层。将有机相水洗,用无水Na2SO4干燥过夜,浓缩。残余物用丙酮溶解,维持反应温度15-20℃下滴加琼斯试剂(Jones),继续反应。抽滤,滤液浓缩,残余物用水溶解,氯仿萃取,并水洗,无水Na2SO4干燥,浓缩得到式(IV)的化合物5-甲基-4-丙酮基-1,3-二氧杂环戊烯-2-酮,用气相色谱(GC)控制纯度>95%。
步骤2:
依次将DMF、式(V)的化合物6,7-二氟-1-甲基-4-氧代-4H-(1,3)硫氮杂环丁烷并(3,2-a)-喹啉-3-羧酸乙酯(作为起始原料)投入反应三口瓶中,再加入式(VI)的无水哌嗪,搅拌反应。过滤,乙醇洗涤,收集固体,干燥,得到浅黄色固体产物,备用。依次将叔丁醇、水、KOH投入三口瓶,搅拌溶解后,加入上述浅黄色固体产物。在55~60℃下搅拌反应。冷却,当物料温度低于30℃时,加入冰水,用盐酸调PH值到6.5~7.0,有固体析出,过滤,水洗,收集固体。干燥,得到浅黄色固体的式(VII)的化合物6-氟-1-甲基-4-氧代-7-(1-哌嗪基)-4H-(1,3)硫氮杂环丁烷(3,2-a)并喹啉-3-羧酸(熔点:208~215℃,水分:9~12%,HPLC控制纯度>98.5%)。
步骤3:
将乙腈投入到三口瓶中,控制室温,并依次加入式(IV)的化合物5-甲基-4-丙酮基-1,3-二氧杂环戊烯-2-酮、式(VII)的化合物6-氟-1-甲基-4-氧代-7-(1-哌嗪基)-4H-(1,3)硫氮杂环丁烷(3,2-a)并喹啉-3-羧酸、以及醋酸钯,搅拌反应,HPLC跟踪反应,控制式(VII)的化合物剩余5%以下,终止反应。滤去固体,将清液倒入搅拌下的水中,用盐酸调节PH值为6~7,固体析出。过滤,固体再以乙醇浸泡后,过滤,干燥,获得式(I)的普卢利沙星粗品(HPLC控制纯度>96%)。将该粗品、活性炭悬浮在乙腈中回流,热过滤,冷却析晶,从而获得普卢利沙星产品(HPLC纯度99.5%)。
参考文献
[1][中国发明,中国发明授权]CN200510037438.8普卢利沙星的制备方法
[2][中国发明,中国发明授权]CN201210139610.0一种普卢利沙星的制备方法
[3][中国发明,中国发明授权]CN200810187274.0合成普卢利沙星的新方法



欢迎您浏览更多关于普卢利沙星的相关新闻资讯信息