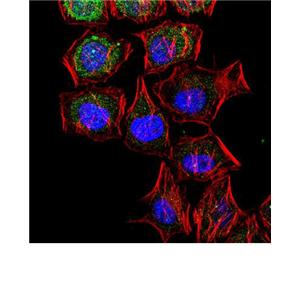
产品概述
| 产品名称(Product Name) | OCRL Rabbit Polyclonal Antibody |
| 描述(Description) | Rabbit Polyclonal Antibody |
| 宿主(Host) | Rabbit |
| 应用(Application) | WB,ELISA |
| 种属反应性(Reactivity) | Human,Mouse |
产品性能
| 偶联物(Conjugation) | Unconjugated |
| 修饰(Modification) | Unmodified |
| 同种型(Isotype) | IgG |
| 克隆(Clonality) | Polyclonal |
| 形式(Form) | Liquid |
| 存放说明(Storage) | Store at 4°C short term. Aliquot and store at -20°C long term. Avoid freeze/thaw cycles. |
| 储存溶液(Buffer) | Liquid in PBS containing 50% glycerol, 0.5% BSA and 0.02% New type preservative N. |
| 纯化方式(Purification) | Affinity purification |
免疫原
| 基因名(Gene Name) | OCRL |
| 别名(Alternative Names) | OCRL; INPP5F; OCRL1; Inositol polyphosphate 5-phosphatase OCRL-1; Lowe oculocerebrorenal syndrome protein |
| 基因ID(Gene ID) | 4952 |
| 蛋白ID(SwissProt ID) | Q01968 |
产品应用
| 稀释比(Dilution Ratio) | WB 1:500-1:2000, ELISA 1:10000.Not yet tested in other applications. |
| 蛋白分子量(Molecular Weight) | 104kDa |
研究背景
This gene encodes an inositol polyphosphate 5-phosphatase. This protein is involved in regulating membrane trafficking and is located in numerous subcellular locations including the trans-Golgi network, clathrin-coated vesicles and, endosomes and the plasma membrane. This protein may also play a role in primary cilium formation. Mutations in this gene cause oculocerebrorenal syndrome of Lowe and also Dent disease. Alternate splicing results in multiple transcript variants. [provided by RefSeq, Jan 2016],catalytic activity:1-phosphatidyl-1D-myo-inositol 4,5-bisphosphate + H(2)O = 1-phosphatidyl-1D-myo-inositol 4-phosphate + phosphate.,caution:It is uncertain whether Met-1, Met-18 or Met-20 is the initiator.,disease:Defects in OCRL are the cause of Dent disease type 2 (DD2) [MIM:300555]. DD2 is a renal disease belonging to the 'Dent disease complex', a group of disorders characterized by proximal renal tubular defect, hypercalciuria, nephrocalcinosis, and renal insufficiency. The spectrum of phenotypic features is remarkably similar in the various disorders, except for differences in the severity of bone deformities and renal impairment. Characteristic abnormalities include low-molecular-weight proteinuria and other features of Fanconi syndrome, such as glycosuria, aminoaciduria, and phosphaturia, but typically do not include proximal renal tubular acidosis. Progressive renal failure is common, as are nephrocalcinosis and kidney stones.,disease:Defects in OCRL are the cause of Lowe syndrome [MIM:309000]; also known as Lowe oculocerebrorenal syndrome. The Lowe syndrome is an X-linked multisystem disorder affecting eyes, nervous system, and kidney. It is characterized by hydrophthalmia, cataract, mental retardation, vitamin D-resistant rickets, aminoaciduria, and reduced ammonia production by the kidney. Ocular abnormalities include cataract, glaucoma, microphthalmos, and decreased visual acuity. Developmental delay, hypotonia, behavior abnormalities, and areflexia are also present. Renal tubular involvement is characterized by impaired reabsorption of bicarbonate, amino acids, and phosphate. Musculoskeletal abnormalities such as joint hypermobility, dislocated hips, and fractures may develop as consequences of renal tubular acidosis and hypophosphatemia. Cataract is the only significant manifestation in carriers and is detected by slit-lamp examination.,function:Converts phosphatidylinositol 4,5-bisphosphate to phosphatidylinositol 4-phosphate. Also converts inositol 1,4,5-trisphosphate to inositol 1,4-bisphosphate and inositol 1,3,4,5-tetrakisphosphate to inositol 1,3,4-trisphosphate. May function in lysosomal membrane trafficking by regulating the specific pool of phosphatidylinositol 4,5-bisphosphate that is associated with lysosomes.,similarity:Belongs to the inositol-1,4,5-trisphosphate 5-phosphatase type II family.,similarity:Contains 1 Rho-GAP domain.,tissue specificity:Brain, skeletal muscle, heart, kidney, lung, placenta and fibroblasts.,
研究领域
Inositol phosphate metabolism;Phosphatidylinositol signaling system;