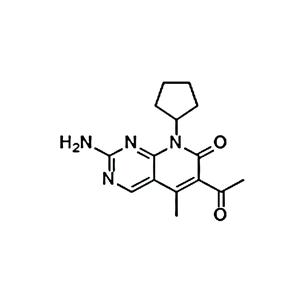
| 名称 | Cebranopadol |
| 描述 | Cebranopadol (GRT6005) is an analgesic NOP and opioid receptor agonist with Kis of 0.9 nM, 0.7 nM, 2.6 nM, 18 nM for human NOP, μ-opioid (MOP), κ-opioid (KOP) and δ-opioid (DOP) receptor, respectively. |
| 激酶实验 | Rat MOP, KOP, and NOP receptor binding assays were run using membrane suspensions from rat brain without the cerebellum for MOP receptors; without the pons, medulla oblongata, and cerebellum for NOP receptors; and without the pons, medulla oblongata, cerebellum, and cortex for KOP receptors and the following tritium-labeled radioligands: [3H]DAMGO in the MOP receptor assay, [3H]nociceptin in the NOP receptor assay, and [3H]Ci-977 in the KOP receptor assay. The assay buffer used for the binding studies was 50 mM Tris-HCl (pH 7.4) supplemented with 0.05% sodium azide. The final assay volume of 250 μl/well included 2 nM [3H]DAMGO, 1 nM [3H]nociceptin, or 1 nM [3H]Ci-977 as a ligand in the MOP, NOP, or KOP receptor assays, respectively, and cebranopadol in dilution series. Cebranopadol was diluted with 25% DMSO in water to yield a final 0.5% DMSO concentration, which also served as a respective vehicle control. The assays were started by the addition of the membrane suspensions and, after short mixing, the assays were run for 90 minutes at room temperature. All incubations were run in triplicate and terminated by rapid filtration under mild vacuum and two washes of 5 ml of buffer using FP-100 Whatman GF/B filter mats. The radioactivity of the samples was counted after a stabilization and extraction period of at least 15 hours by use of the scintillation fluid Ready Protein; the complete competition curves for cebranopadol were recorded [1]. |
| 动物实验 | The pharmacokinetic properties of cebranopadol in rats were investigated after a single intravenous dose of 160 μg/kg cebranopadol. The intravenous dose was administered as a bolus in a volume of 2 ml/kg with a catheter in the vena femoralis. Blood samples (200 μl/sample) were withdrawn via an implanted arterial catheter (arteria carotis) by an automated blood sampling system at the following sampling times: 0 (predose), 5, 15, 30, 60, 180, 360, 720, and 1440 minutes after administration. Blood samples were centrifuged, and plasma was separated. Plasma concentrations of cebranopadol were determined using a validated liquid chromatography-tandem mass spectrometry method. The lower limit of quantification for cebranopadol in this method was 0.05 ng/ml using a sample volume of 50 μl of plasma [1]. |
| 体外活性 | Cebranopadol在人类MOP和DOP受体上展示了完全激动剂效能,在人类NOP受体上展现了近乎完全的效能,并在人类KOP受体上表现了部分效能。在使用表达人类5-HT5A受体的膜进行的功能性[35S]GTPgS结合实验中,Cebranopadol在高达10.0 μM的浓度下既未显示激动剂效应也未表现出显著的拮抗效应[1]。 |
| 体内活性 | Cebranopadol在多种大鼠急性和慢性痛模型中显示出高效和高效能的抗痛觉过敏和抗高敏感性效果,经静脉注射后的ED50值为0.5-5.6μg/kg,口服后为25.1μg/kg。Cebranopadol的作用持续时间长(静脉注射12μg/kg后达7小时;口服55μg/kg后在大鼠尾巴抽动测试中超过9小时)[1]。在streptozotocin(STZ)处理的大鼠中,Cebranopadol(i.pl.)减轻了同侧爪的机械性高敏感,但在对侧爪无效果。在CCI大鼠中,Cebranopadol(i.pl.)在同侧爪显示出抗痛觉丧失活性。向对侧爪注射后,Cebranopadol也展现了同侧的抗痛觉丧失活性,但效力降低且起效延迟。在糖尿病小鼠中,Cebranopadol i.th.和i.c.v.以完全有效性和类似的效力减少了热性高痛敏感性[2]。在NOP(-/-)小鼠中,吗啡处理产生了与NOP(+/+)动物相同的撤药症状,而Cebranopadol处理在NOP(-/-)小鼠中引起了比NOP(+/+)小鼠更强烈的撤药综合症[3]。 |
| 存储条件 | Powder: -20°C for 3 years | In solvent: -80°C for 1 year | Shipping with blue ice. |
| 溶解度 | DMSO : 5 mg/mL (13.21 mM), Sonication is recommended.
H2O : Insoluble
|
| 关键字 | Inhibitor | inhibit | Opioid Receptor | GRT-6005 | Cebranopadol | GRT 6005 |
| 相关产品 | Docusate sodium | Progesterone | Naltrexone hydrochloride | SCH 221510 | Matrine | Amentoflavone | Mirtazapine | Mianserin hydrochloride | (-)-Menthol | Bevenopran | Naloxone HCl Dihydrate | Trimebutine |
| 相关库 | 经典已知活性库 | 已知活性化合物库 | GPCR靶点分子库 | 临床期小分子药物库 | 膜蛋白靶向化合物库 | 神经退行性疾病化合物库 | 疼痛相关化合物库 | 药物功能重定位化合物库 | 抗癌临床化合物库 | 抗癌药物库 |