化合物 Quinidine Monosulfate|T61517|TargetMol
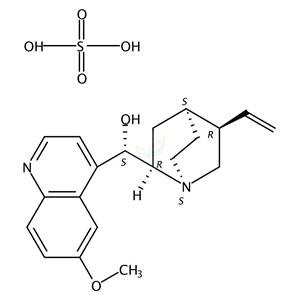
Quinidine Monosulfate
50-54-4
50-54-4
¥19420
50mg
起订
¥24625
100mg
起订
¥14900
25mg
起订
上海 更新日期:2026-06-29
产品详情:
- 中文名称:
- 化合物 Quinidine Monosulfate
- 英文名称:
- Quinidine Monosulfate
- CAS号:
- 50-54-4
- 品牌:
- TargetMol
- 产地:
- 美国
- 保存条件:
- Powder: -20°C for 3 years | In solvent: -80°C for 1 year Shipping with blue ice/Shipping at ambient temperature.
- 产品类别:
- 抑制剂
- 货号:
- T61517
公司简介
TargetMol Chemicals Inc. 总部位于马萨诸塞州波士顿,致力于为全球生化领域科学家的研究提供专业的产品和服务。TargetMol?品牌的客户群分布于40多个国家和地区,已发展成为全球知名的化合物库和小分子化合物研究供应商。 TargetMol?可提供160多种满足不同需求的化合物库,以及多种类型的生化试剂产品,包括12000多种抑制剂、16000多种天然产物和各类多肽、抗体、生命科学试剂盒等,此外,我们还建设有CADD(计算机辅助药物设计)研究中心、药理实验室、药化合成平台三大技术中心,全方位满足客户的定制需求。 凭借我们优质的产品和服务、快速高效的全球供应链和专业的技术支持,我们将有效帮助您缩短研发周期,取得更成功的结果。
| 成立日期 | (14年) |
| 注册资本 | 566.265100万人民币 |
| 员工人数 | 100-500人 |
| 年营业额 | ¥ 1亿以上 |
| 经营模式 | 贸易,工厂,试剂,定制,服务 |
| 主营行业 | 天然产物,生化试剂,分子生物学,分子砌块,生物技术服务 |
化合物 Quinidine Monosulfate相关厂家报价 更多
-

- 硫酸奎尼丁 50-54-4 医药中间体 标准品 纯度≥99% COA随货
- 湖北拓源精细化工有限公司 VIP
- 2026-07-31
- ¥5500
-
- 硫酸奎尼丁 Quinidine sulfate
- 成都彼样生物科技有限公司 VIP
- 2026-07-30
- 询价
-

- 硫酸奎尼丁
- 武汉维斯尔曼生物工程有限公司 VIP
- 2026-07-30
- 询价
-

- 硫酸奎尼丁—50-54-4
- 湖北魏氏化学试剂股份有限公司 VIP
- 2026-07-27
- ¥588
-

- 硫酸奎尼丁
- 湖北威德利化学科技有限公司 VIP
- 2026-07-25
- ¥9000
-

- 硫酸奎尼丁—50-54-4
- 湖北魏氏化学试剂股份有限公司 VIP
- 2026-07-21
- ¥1350
-

- 50-54-4;硫酸奎尼丁
- 普善实业(陕西)有限公司 VIP
- 2026-06-26
- 询价
-
- 硫酸奎尼丁 50-54-4
- 陕西西化化学工业有限公司 VIP
- 2026-05-25
- 询价
-

- 硫酸奎尼丁
- 广州千行进出口有限公司
- 2023-11-21
- 询价
-

- 硫酸奎尼丁
- 湖北麦凯斯精化科技有限公司
- 2023-03-23
- 询价
