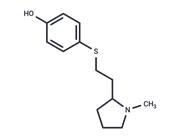
| Name | EZM 2302 |
| Description | EZM 2302 (GSK3359088) is a selective, and orally available arginine methyltransferase CARM1 inhibitor (IC50: 6 nM).[1] |
| Cell Research | Cultured cells in linear/log phase growth were split to a seeding density of 2e5 cells/mL in 2–20mLs of media, depending on the yield required at the end of the growth period. The compound was diluted in DMSO and added to each culture vessel with a final DMSO concentration of 0.2%. Cells were allowed to grow for 96?hours. At the conclusion of each treatment period, cells were harvested by centrifugation (5?minutes, 1200?rpm), and cell pellets were rinsed once with PBS before being frozen on dry ice pending further processing. |
| Kinase Assay | CARM1 activity was measured as previously reported for the histone methyltransferases PRMT1/6/865 and PRMT558. Briefly, CARM1 was preincubated with compounds for 30?minutes at room temperature before reactions were initiated. Final assay conditions were 0.25?nM CARM1, 30?nM 3H-S-adenosyl-methionine (SAM), and 250?nM biotinylated peptide in buffer containing 20?mM bicine, 1?mM tris(2-carboxyethyl)phosphine, 0.005% bovine skin gelatin and 0.002% Tween-20, pH 7.5. The assays were quenched by the addition of 300?μM unlabeled SAM. The quantity of 3H-labeled peptide produced was measured by Flashplate. |
| Animal Research | For the in vivo efficacy studies, there were 8 mice per dose group and each mouse was inoculated subcutaneously at the right flank. All cells were suspended in a 0.2?mL mixture of base media and Matrigel at 1:1 for tumor development. RPMI-8226 cells were inoculated at 5?×?10^6 cells/mouse and treatment began when the mean tumor sizes reached 120?mm3 (28 days post-inoculation). CB-17 SCID Mice were assigned into groups using a randomized block design. EZM2302 or vehicle (0.5% methylcellulose in water) was administered orally BID at a dose volume of 37.5, 75, 150, or 300?mg/kg for 21 days. Body weights were measured twice a week for the duration of the study. Tumor size was measured twice weekly in two dimensions using a caliper, and the volume was expressed in cubic millimeters. Animals were euthanized 3?hours post-final dose, with blood and tissues collected for analysis. |
| In vitro | METHODS: The effects of EZM 2302 (GSK3359088) (0.3/1/5/20/80/300/1250/5000nM) treatment on cellular methylation were tested by immunoblotting in the multiple myeloma (MM) cell line RPMI-8226.
RESULTSEZM 2302 (GSK3359088) inhibited cellular PABP1 and SMB methylation in vitro. [1] |
| In vivo | METHODS: Male CD-1 mice and male Sprague-Dawley rats (n = 3) were treated with a single dose of EZM 2302 (GSK3359088) at 2 mg/kg by intravenous injection (i.v.) and 10 mg/kg by oral gavage (p.o.; mice only), and another group of rats cannulated in the jugular and portal veins were dosed by oral gavage (10 mg/kg in 0.5% methylcellulose in water). Approximately 110 μL of blood was collected from animals at prespecified time intervals by retro-orbital bleeding (mice), caudal vein (i.v. in rats), or jugular and portal vein sampling (p.o. in rats). The 2-hour samples were split for parallel determination of blood and plasma concentrations (ex vivo ratio).
RESULTS EZM 2302 (GSK3359088) was stable in human hepatocytes (CL <3 mL/min/kg) and modestly bound to human, mouse, and rat plasma proteins with mean unbound fractions of 0.66, 0.46, and 0.74, respectively. In mice and rats, plasma clearance (CL) was 43 and 91 mL/min/kg, respectively (Table 1 Fig. 4a, b). In rats, low levels of binding to erythrocytes were observed, while EZM2302 showed no blood partitioning in mice; therefore, the blood CL was equivalent in both species. Although rats showed an intermediate mean bioavailability (F), the proportion of dose absorbed from the gastrointestinal tract (Fa*Fg) was much higher at 81%, as measured by the JVC-PVC rat PK study, reflecting the high permeability of EZM 2302 (GSK3359088). Therefore, EZM2302 has oral bioavailability and is suitable for in vivo studies. [1]
METHODS: CB-17 SCID mice were assigned to groups using a randomized group design. EZM 2302 (GSK3359088) or vehicle (0.5% methylcellulose in water) was orally administered BID for 21 days at doses of 37.5, 75, 150, or 300 mg/kg. Body weight was measured twice weekly during the study. Tumor size was measured twice weekly in two dimensions using a caliper. Animals were euthanized 3 hours after the last dose, and blood and tissues were collected for analysis.
RESULTS In the RPMI-8226 xenograft model, EZM2302 showed dose-dependent exposure and tumor growth inhibition after 21 days; tumors in all EZM 2302 (GSK3359088) dose groups measured on day 21 showed significant reductions in tumor growth compared to vehicle, with tumor growth inhibition ranging from 45% in the 37.5 mg/kg dose group to 63% in the 300 mg/kg dose group; RPMI-8226 xenograft tumors collected on day 21 showed a dose-dependent decrease in methylation of all tested CARM1 substrates, and a statistically significant increase in unmethylated SmB (SmBme0) was detected in all dose groups, from 8-fold at 37.5 mg/kg to 14-fold at 150 mg/kg. aDMA levels were also significantly reduced at all dose groups, with a maximum inhibition of 65% observed at the 75 mg/kg dose group. Levels of total and methylated PABP1 are difficult to detect in xenograft tissues.[1] |
| Storage | Powder: -20°C for 3 years | In solvent: -80°C for 1 year | Shipping with blue ice. |
| Solubility Information | DMSO : < 1 mg/mL (insoluble)
|
| Keywords | GSK 3359088 | inhibit | Inhibitor | GSK-3359088 | Histone Methyltransferase | EZM 2302 | EZM2302 | EZM-2302 |
| Inhibitors Related | BIX-01294 trihydrochloride | Tazemetostat | Tazemetostat hydrobromide | Piribedil | XY1 | UNC 0631 | GSK126 | MAK683 | EPZ015666 | AMI-1 free acid | MS37452 | MRTX-1719 |
| Related Compound Libraries | Histone Modification Compound Library | Bioactive Compound Library | Epigenetics Compound Library | Chromatin Modification Compound Library | Inhibitor Library | NO PAINS Compound Library | Orally Active Compound Library | Bioactive Compounds Library Max |

 United States
United States